
What It Means for FDA Operations and the Diagnostics Industry
Authored by: Beaufort Contributor
When the federal government shut down on October 1, 2025, the U.S. Food and Drug Administration (FDA) scaled back operations, focusing on authorized and essential public health functions. While the FDA remains open, its work is now limited to activities necessary to ensure public health and safety and those funded by carryover user fees — funds collected in prior fiscal years that remain available for use.
FDA staff continue to work on ongoing safety monitoring and active submission reviews, but most new submissions and longer-term initiatives are on hold until Congress passes a funding bill.
The official contingency plan from the U.S. Department of Health and Human Services (HHS) outlines how the FDA will operate during this lapse in appropriations.
You can view it here:
https://www.hhs.gov/about/budget/fy-2026-hhs-contingency-staffing-plan/index.html
FDA Priorities During the Lapse
The FDA continues to function with a reduced workforce — about 86% of its staff — consisting of:
- Exempt employees, whose work is supported by carryover user-fee funding or other existing resources.
- Excepted employees, whose duties are considered essential to protecting life or property.
These teams sustain FDA’s mission-critical operations: overseeing product safety, managing recalls, monitoring imports, and responding to urgent public health risks.
FDA Activities That Continue
During the shutdown, FDA continues the functions directly tied to immediate health and safety and those sustained by carryover user-fee balances. Carryover balances vary by program, meaning review activity may continue longer in some divisions than others. These include:
- Monitoring and responding to urgent safety threats, including product recalls and disease outbreaks.
- Conducting import surveillance and other product safety monitoring activities.
- Continuing review of existing premarket applications for drugs, biologics, and medical devices that were submitted and paid for before October 1.
- Conducting for-cause inspections and limited surveillance inspections.
- Maintaining oversight of ongoing clinical trials where participant safety could be at risk.
For diagnostic developers, this means premarket submissions already under review — such as 510(k)s, PMAs, or De Novo Classification Requests — may continue moving forward if previously funded, though applicants may experience a slower pace due to any reductions in staffing.
FDA Activities on Hold — and Why It Matters
Most other FDA activities are paused until Congress passes a new funding bill. These include:
- Acceptance and processing of new premarket submissions that require user-fee payment (e.g., new 510(k)s, PMAs, or De Novo Classification Requests).
- Scheduling of new advisory committee meetings or long-term policy development.
- Routine, non-critical inspections of manufacturing facilities.
- Hiring, staff training, and other internal improvement programs.
The FDA’s contingency plan also notes that regulatory science research and program development activities will pause. In practical terms, this means:
- Internal research that supports future policy or technical guidance (for example, performance standard development or model validation work) is temporarily halted.
- Upgrades to information technology systems, laboratories, and other infrastructure are postponed.
- Staff development, onboarding, and training are suspended.
While these slowdowns may not affect premarket reviews immediately, they can delay future guidance documents, modernization projects, and cross-agency initiatives — creating ripple effects that last beyond the shutdown itself.
What This Means for Diagnostic Developers
For diagnostic developers, the degree of impact will depend on both the type of submission and the timing of review activity relative to the funding lapse.
Ongoing Premarket Submission Reviews
If your diagnostic premarket application was already under review before October 1 and supported by an existing user-fee payment, FDA reviewers may continue their reviews.
However, applicants should plan for slower communication, longer response times, and possible scheduling changes if FDA resources are limited.
Q-Submissions (Pre-Submissions) and Investigational Device Exemption (IDE) Submissions
Q-Submissions (e.g., pre-submissions) and IDE submissions do not require user fees and therefore may still be accepted and reviewed, though timing may vary depending on available staff and competing priorities. As with premarket review submissions, the FDA’s ability to review these may be constrained during a funding lapse, and sponsors are advised to plan accordingly.
Maintaining Progress During the Shutdown
Even within these constraints, diagnostic sponsors can use this time effectively to maintain readiness and reduce delays when operations resume:
- Address outstanding FDA questions on submissions under review to maintain momentum.
- Refine planned documentation to ensure data packages, validation reports, and labeling are complete.
- Verify internal quality systems and maintain inspection readiness.
- Communicate realistic expectations to internal teams, partners, and other stakeholders regarding potential delays.
When Operations Resume
Once Congress enacts new appropriations, FDA will begin restoring full operations.
Historically, the FDA takes several weeks to re-engage fully, as review divisions re-prioritize pending work and reestablish schedules.
Sponsors can expect:
- Review clocks to restart.
- Rescheduling of postponed meetings or consultations.
- Gradual resumption of policy, guidance, and modernization initiatives.
Being prepared to re-engage quickly helps sponsors regain momentum once the FDA is fully operational.
How Beaufort Can Help
At Beaufort, we understand the pressures a funding lapse places on diagnostics developers — especially when review timelines, study initiation, and market planning depend on FDA availability.
Our team continues to work alongside clients during this period to:
- Assess regulatory impacts and adjust project timelines.
- Prepare and quality-check submissions so they are complete and ready for prompt filing once FDA operations resume.
- Strengthen documentation, validation, and QMS records to ensure post-shutdown readiness.
- Support ongoing clinical and regulatory activities that remain active during the lapse, including IDE oversight and study monitoring.
Our goal is to help sponsors maintain progress, minimize disruption, and be positioned to move forward the moment the FDA reopens.
Conclusion
The FY 2026 government shutdown has not closed the FDA, but it has limited the Agency’s ability to operate as usual.
Critical safety and review activities continue, but most new submissions, guidance development, and long-term policy work are paused until appropriations return.
For diagnostic developers, this period calls for focus and preparation. By maintaining documentation, communication, and compliance readiness now, sponsors can move quickly when the regulatory environment stabilizes.

A new PCCP draft guidance outlines what elements the agency expects in these plans
As published in Today’s Clinical Lab
The U.S. Food and Drug Administration (FDA) recently issued a second draft guidance on predetermined change control plans (PCCPs) for medical devices that could influence how clinical labs modify laboratory-developed tests (LDTs).
PCCPs are documents included in a marketing submission that:
- Describe anticipated changes to the FDA-approved or -cleared medical device;
- Detail how these changes will be implemented and validated.
If approved, these plans allow changes without necessitating a new FDA submission, potentially accelerating a device’s modification and saving FDA review time.
Relevant to devices—including in vitro diagnostic devices (IVDs) and device-led combination products that are reviewed through the 510(k), De Novo, and pre-market approval pathways—the FDA guidance proposes a policy for PCCPs and provides recommendations on the information to include in a PCCP.
The guidance may be of interest to the clinical laboratory community, as PCCPs may offer a way to introduce flexibility for modifying certain LDTs under the FDA’s recently issued final rule on regulating these assays.
Elements of a predetermined change control plan
A PCCP should consist of:
- Description of modifications, which outline the specific, planned changes that may be made to the device, including the specifications for the characteristics and performance of the planned modifications.
- Modification protocol, which describes verification and validation activities, including predefined acceptance criteria, that will support each modification to ensure the device remains safe and effective (or substantially equivalent) across the intended use populations.
- Impact assessment, which identifies the benefits and risks introduced by the specified, planned modifications and addresses how the verification and validation activities of the modification protocol will continue to ensure the safety and effectiveness (or substantial equivalence) of the device.
Here are the key takeaways about the FDA’s guidance on PCCPs:
- Scope: Unlike the FDA’s April 2023 draft guidance that focused on AI- and machine-learning-enabled devices, the new draft clarifies that PCCPs apply broadly, not just to these technologies.
- Purpose: The goal of a PCCP is to provide a least burdensome option for manufacturers to implement device modifications without needing to submit a new marketing submission for each modification—all while maintaining a reasonable assurance of safety and effectiveness (or substantial equivalence).
- Specificity: The modifications included in a PCCP must maintain the device within its intended use and should include specific changes that can be verified and validated.
- Authorization: To gain authorization, a PCCP must provide enough detail to allow the FDA to assess the reasonable assurance of safety and effectiveness (or substantial equivalence).
- Implementation: A PCCP is part of the device’s marketing authorization; therefore, the manufacturer is required to implement modifications consistent with their authorized PCCP.
In general, the FDA’s guidance documents do not establish legally enforceable responsibilities. Instead, guidance describes the agency’s current thinking on a topic and should be viewed as recommendations. The updated draft guidance on PCCPs is open for public comment for 90 days following its official issuance on August 22, 2024.
Learn more about our regulatory services or contact us today to schedule an introductory meeting.
6 Strategies to Help You Meet FDA Requirements and Maintain Market Access
On April 29, 2024, the US Food and Drug Administration (FDA) issued a long-anticipated final rule asserting its authority to regulate laboratory-developed tests (LDTs) as medical devices (“Medical Devices; Laboratory Developed Tests” (89 FR 37286) (“LDT Final Rule”)). This rule, effective May 6, 2024, marks a significant shift from the FDA’s previous enforcement discretion for LDTs, impacting stakeholders across the diagnostics industry.
Below are key highlights of the final rule and how laboratories offering LDTs may begin preparing to meet the new requirements and timelines.
Redefining LDTs as In Vitro Diagnostic (IVD) Medical Devices
The FDA’s final rule makes explicit that in vitro diagnostic products are medical devices under the Federal Food, Drug, and Cosmetic Act (FD&C Act; 21 CFR § 809.3) “including when the manufacturer of the IVD is a laboratory”. While the laboratory community views LDTs as laboratory testing services, FDA defines LDTs as IVDs that are intended for clinical use and that are designed, manufactured, and used within a single laboratory that is certified under the CLIA and meets the regulatory requirements under CLIA to perform high complexity testing.
Phaseout of Enforcement Discretion
Over the next four years, the FDA will phase out its enforcement discretion for most LDTs. By May 6, 2028, all IVDs offered as LDTs must comply with FDA regulations akin to those for other FDA-regulated medical devices. This includes implementing robust quality management systems, adhering to rigorous reporting practices, fulfilling establishment registration and device listing obligations, and undergoing FDA premarket review where applicable. The phased approach aims to provide laboratories with time to adjust to the new regulatory landscape while ensuring patient safety and maintaining access to accurate diagnostic testing.
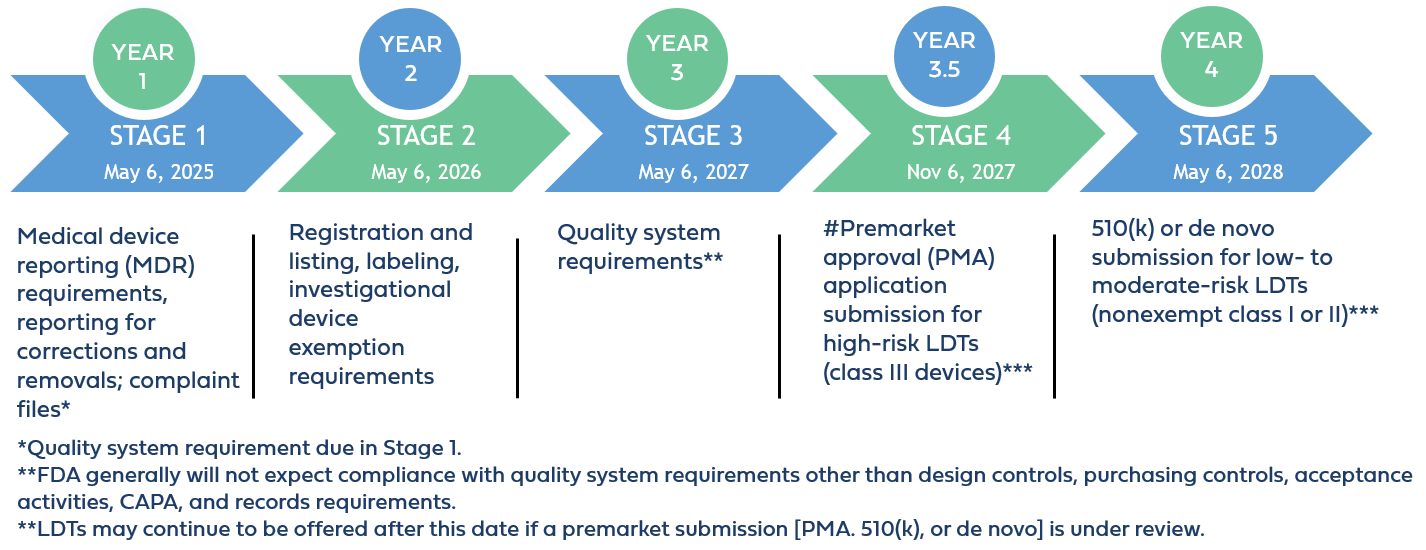
The phaseout policy applies to “IVDs offered as LDTs” defined by the FDA as IVDs that are manufactured and offered as LDTs by laboratories that are certified to perform high complexity testing under CLIA, and used within such laboratories, even if those IVDs do not fall within FDA’s traditional understanding of an LDT because they are not designed, manufactured, and used within a single laboratory.
Implementation Stages and Requirements
STAGE 1: Medical Device Reporting Requirements
Beginning on May 6, 2025, which is 1 year after the publication date of the final LDT rule, laboratories offering IVDs as LDTs must comply with Stage 1 requirements, which include medical device reporting requirements (21 CFR part 803), corrections and removals (21 CFR part 806), and quality system (QS) requirements for establishing and maintaining complaint files (21 CFR 820.198).
These requirements will be new to laboratories because they are distinct from those under Clinical Laboratory Improvement Amendments (CLIA) (42 CFR Part 493). While CLIA mandates procedures for managing complaints related to the quality of laboratory testing services, the FDA’s requirements under 21 CFR 820.198 specifically pertain to managing complaints related to medical devices. This includes establishing and maintaining procedures to receive, review, and evaluate complaints alleging device deficiencies.
STAGE 2: Registration and Listing, Labeling, and Investigational Device Exemption Requirements
Beginning on May 6, 2026, laboratories offering IVDs as LDTs must comply with Stage 2 requirements. These encompass establishment registration and device listing obligations (510 of the FD&C Act, 21 CFR part 607, and 21 CFR part 807 (excluding subpart E)), labeling requirements (section 502 of the FD&C Act and 21 CFR parts 801 and 809, subpart B); and investigational device exemption requirements (section 520(g) of the FD&C Act and 21 CFR part 812).
Compliance will demand understanding and adherence to FDA specifications for labeling, including formatting and content stipulations under 21 CFR parts 801 and 809, as well as ensuring the inclusion of Unique Device Identifiers (UDIs) on labels for traceability throughout the device lifecycle. Establishing a relationship with a UDI issuing agency is necessary to obtain UDIs and assign them to each device model or version, ensuring that the identifier contains essential information such as device identifier, production identifier, and, where applicable, lot or serial number.
Laboratories must also adhere to FDA’s regulatory framework for investigational use of medical devices, when IVDs offered as LDTs are used in clinical investigations. This will include determining the appropriate IVD risk (significant, non-significant, or IDE exempt), obtaining necessary study approvals, and ensuring ongoing compliance throughout the investigation.
STAGE 3: Quality System Regulations (QSR) Implementation
Beginning on May 6, 2027, laboratories offering IVDs as LDTs must comply with Stage 3 requirements, which encompass specific QSR requirements outlined in 21 CFR 820. These include implementing design controls, purchasing controls, and corrective and preventive actions (CAPA).
On February 2, 2024, FDA issued a final rule amending the device QS regulation, 21 CFR part 820, to align more closely with international consensus standards for devices (89 FR 7496, available at https://www.federalregister.gov/d/2024-01709).When the final rule takes effect, FDA will also update the references to provisions in 21 CFR part 820 in this guidance to be consistent with that rule.
Key Components of Design Controls: Central to QSR compliance are Design Controls, a structured approach to product development that ensures devices meet specified requirements and are safe and effective for their intended use. Laboratories developing IVDs as LDTs must establish and maintain processes and procedures that encompass the following key components:
- Design and Development Planning (DDP): Laboratories will be required to create a comprehensive DDP outlining the design control activities and milestones for each device. This plan defines roles and responsibilities of team members involved in the design process and sets out a clear roadmap for development.
- Design Inputs: Laboratories will be required to translate user needs and intended use into specific design inputs. Design inputs encompass functional, performance, and safety requirements that the device must meet to fulfill its intended purpose. These inputs serve as the foundation for subsequent design activities.
- Design Outputs: Laboratories must generate detailed design outputs that specify device characteristics, including drawings, specifications, and other relevant documentation. Design outputs must demonstrate the design meets the design inputs and must be meticulously documented to ensure traceability and compliance.
- Design Reviews: Laboratories must conduct systematic design reviews at critical stages of development to evaluate progress, identify any issues or shortcomings, and verify alignment with design requirements. These reviews ensure that potential problems are identified early and addressed promptly to maintain project timelines and product quality.
- Design Verification: Verification involves rigorous testing and analysis to confirm that the device functions as intended. This process ensures that design outputs meet the specified design inputs and are validated through objective evidence. Verification activities must be well-documented and include testing protocols, results, and conclusions.
- Design Validation: Validation is conducted under defined operating conditions to ensure that the final device meets user needs and intended use. This stage involves comprehensive testing in simulated or actual use environments, including software validation and risk analysis where applicable. Validation activities provide assurance that the device is safe, effective, and suitable for its intended clinical application.
- Risk Management: Throughout the design process, laboratories must implement a systematic approach to identify, analyze, control, and monitor risks associated with the device throughout the design process in compliance with the current version of International Organization for Standardization (ISO) 14971.
- Design Transfer: Ensures a smooth transition of the approved design to production through design transfer activities. This stage includes verification that production processes can consistently produce devices that meet design specifications.
- Design Changes: Laboratories must establish procedures for managing design changes. throughout the device lifecycle. Evaluation, documentation, and validation of design changes are required before implementation to prevent unintended alterations that could compromise device safety, efficacy, or intended use. Any modifications must be carefully controlled and documented to maintain regulatory compliance.
STAGE 4: Premarket Review for High Risk IVDs Offered as LDTs
Beginning on November 6, 2027, laboratories offering Class III IVDs as LDTs must comply with Stage 4 requirements, requiring FDA premarket approval (PMA) before marketing. This entails comprehensive analytical and clinical evaluations to demonstrate device safety and effectiveness, aligning with stringent FDA review standards under 21 CFR 814.20. The PMA application must include all elements required under 21 CFR 814.20, unless omissions can be justified by the applicant.
Separately, the FDA’s Center for Devices and Radiological Health (CDRH) intends to initiate the reclassification process for certain IVDs currently classified as Class III (high-risk devices) to Class II (moderate-risk devices). This reclassification would potentially enable these tests to undergo review through the less burdensome premarket notification (510(k)) pathway instead of the premarket approval (PMA) pathway, the most stringent type of FDA medical device review.
However, the FDA will only consider reclassification for Class III devices if the agency has sufficient information (i.e., experience) to establish special controls that, together with general controls, provide a reasonable assurance of safety and effectiveness for these tests. As a result, only those Class III IVDs for which the FDA can establish appropriate special controls will be eligible for down classification to Class II.
STAGE 5: Premarket Review for Moderate and Low Risk IVDs Offered as LDTs
Beginning on May 6, 2028, laboratories offering Class II moderate-risk and Class I low-risk IVDs as LDTs must submit a 510(k) premarket notification demonstrating substantial equivalence to legally marketed devices, unless exempt. This stage also allows for alternative pathways like De Novo classification requests for devices lacking predicate devices but deemed safe and effective under general or special controls.
Additionally, certain other premarket submissions, such as Humanitarian Device Exemption applications or Biologics License Applications (BLAs), may also be appropriate depending on the specific circumstances of the IVD offered as an LDT.
Of note, for IVDs that rely on or otherwise incorporate software (including off-the-shelf software) or that meet the definition of a “cyber device” (Section 524B(c) of the FD&C Act), premarket submissions will need to be inclusive of documentation for FDA’s review and evaluation. For IVDs offered as LDTs already on the market, if a premarket submission has been received by the beginning of Stage 4 or Stage 5 (as applicable), the FDA intends to exercise enforcement discretion during the review period to ensure continued patient access to these devices without interruption.
The agency’s phaseout transition aims to enhance FDA oversight and ensure the safety and effectiveness of LDTs, by aligning expectations for regulatory compliance with that of commercially distributed medical devices. This phased approach is intended to allow laboratories time to comply with the new regulatory framework while continuing to meet the needs of patients and healthcare providers for accurate diagnostic testing.
Notably the amended regulation and phaseout policy do not change other requirements for laboratories, including requirements under the Clinical Laboratory Improvement Amendments of 1988 (CLIA), which are separate from requirements under the FD&C Act.
Continued General Enforcement Discretion Approach for Certain IVDs
The FDA’s LDT Final Rule, in comparison to its proposed rule, broadens the scope of LDTs for which the agency intends to continue exercising general enforcement discretion. This means that the FDA will generally not enforce any applicable requirements for the following:
- 1976-Type LDTs: These tests use manual techniques and are performed by laboratory personnel with specialized expertise and rely on clinical-use (not research use only) components.
- Certain Human Leukocyte Antigen (HLA) Tests for Transplantation: Specifically, histocompatibility testing when used in connection with organ, stem cell, and tissue transplantation to perform HLA allele typing, for HLA antibody screening and monitoring, or for conducting real and “virtual” HLA crossmatch tests.
- Forensic Tests: Tests intended solely for forensic (law enforcement) purposes.
- US Department of Defense (DoD) or the Veterans Health Affairs (VHA) LDTs: LDTs manufactured and performed within the VHA or DoD and used for patients tested and treated within the DoD or VHA.
Tests manufactured and offered for use exclusively for public health surveillance are also not affected by the phaseout policy.
Targeted Enforcement Discretion Approach for Other IVDs
Additionally, the FDA has outlined a targeted enforcement discretion approach for certain other categories of LDTs. Under this approach, the FDA intends not to enforce certain applicable requirements for these IVDs. This decision is based on the FDA’s assessment that tests in these categories are unlikely to pose significant risks, or they are conducted under circumstances that inherently mitigate potential risks. These IVDs offered as LDTs include:
- Certain Modified Versions of Another Manufacturer’s 510(k) Cleared or De Novo Authorized Test
- LDTs Approved by New York State Department of Health’s Clinical Laboratory Evaluation Program (NYSDOH CLEP)
- LDTs for Unmet Needs Manufactured and Performed by Laboratories in an Integrated Healthcare System
- Currently Marketed LDTs (i.e., prior to May 6, 2024) and “Not Significantly Modified”
- Non-Molecular Antisera LDTs for Rare Red Blood Cell (RBC) Antigens for Transfusion Compatibility
The table below provides a high-level summary of the key categories of LDTs under FDA enforcement discretion and the regulatory requirements to which compliance is expected in Stages 1 through 5 of the agency’s phaseout policy.
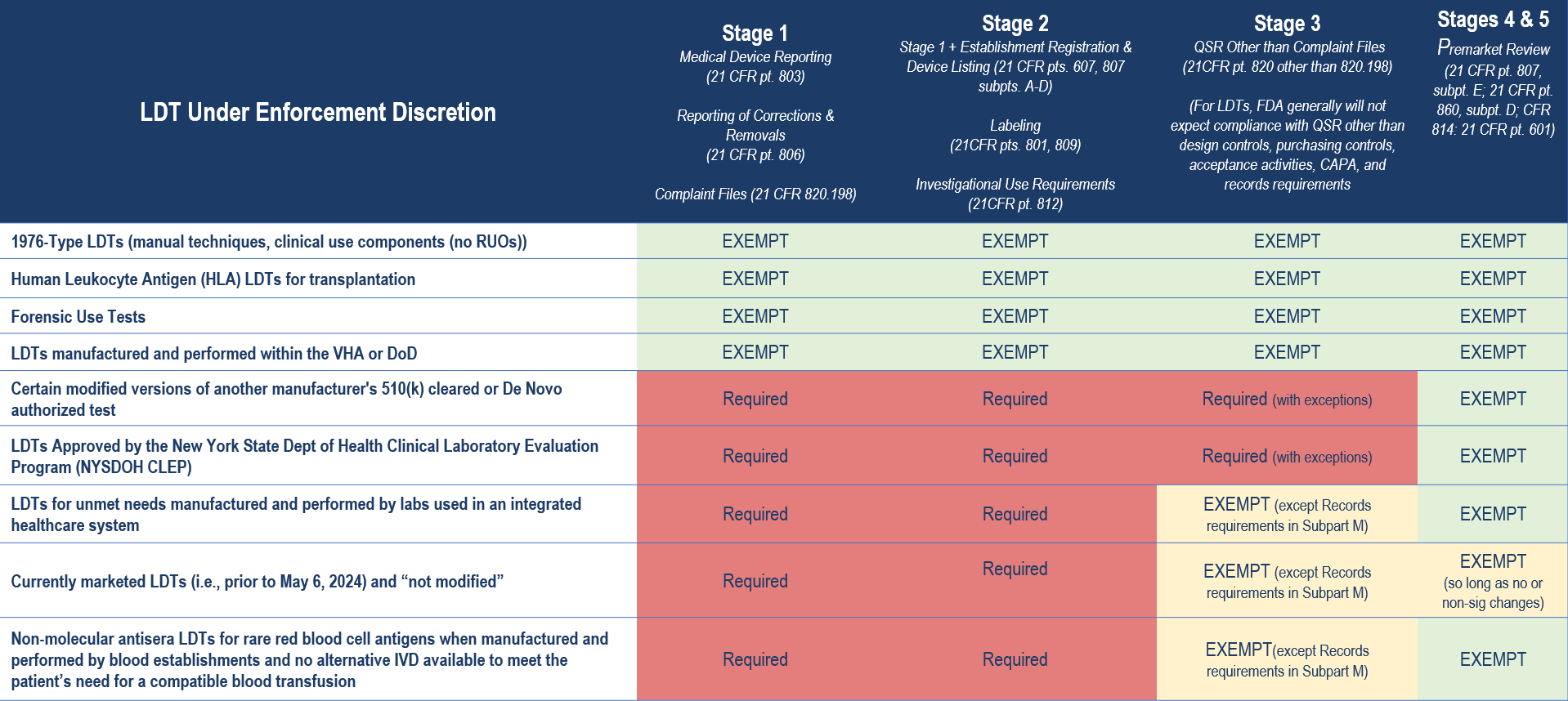
Importantly, there are additional limitations to the agency’s enforcement discretion policies. For example, for LDTs that are modified versions of another manufacturer’s 510(k) cleared or De Novo authorized test, the FDA’s enforcement discretion policy exempting premarket review only applies when the modification was made following design control and other quality system requirements and in a manner that could not “significantly affect the safety or effectiveness of the test and does not constitute a major change or modification in intended use, and where the modified test is performed only in the laboratory making the modification”. Otherwise, FDA expects premarket submissions from laboratories modifying a third party’s 510(k) cleared or De Novo authorized test for the same types of changes for which FDA would expect a premarket submission from the original manufacturer making changes to its own IVD.
Similarly, any “significant modifications” to an LDT marketed before May 6, 2024 will require meeting compliance expectations that align with the phaseout policy. And, for LDTs for unmet needs manufactured and performed by laboratories in an integrated healthcare system, enforcement discretion would end when an FDA-authorized test that addresses the unmet need becomes available.
Moreover, as with any enforcement discretion policy, FDA may update any of these policies as circumstances warrant or if the circumstances that inform these policies change, consistent with FDA’s good guidance practices (section 701(h) of the FD&C Act, 21 CFR 10.115). Additionally, regardless of the phaseout timeline and enforcement discretion policies for certain IVDs, FDA retains discretion to pursue enforcement action for violations of the FD&C Act at any time.
Laboratories will need to determine the FDA’s applicable regulations and enforcement discretion policies that apply to each of their LDTs. This will guide decisions on compliance pathways and documentation requirements and help in determining needed resources.
6 Strategies to Help You Get Started
- Stay Informed
- Stay abreast of legislative developments and regulatory changes that may impact LDT regulation.
- This includes proposed legislation, and changes in enforcement policies as well as FDA announcements, guidance documents, webinars, and other resources.
- Understand FDA Requirements
- Become familiar with FDA definitions, requirements, and expectations for LDTs.
- This includes understanding the differences between LDTs and FDA-cleared/approved tests and knowing when FDA regulation applies.
- Know your Test Status
- Determine the FDA’s applicable regulations and enforcement discretion policies that apply to each type of LDT.
- This will guide decisions on compliance pathways and documentation requirements and help in determining needed resources.
- Quality System Compliance
- Evaluate your laboratory’s CLIA Quality Management System (QMS) against FDA’s Quality System Regulations (QSR) to identify gaps.
- Begin planning necessary changes to align with FDA requirements, ensuring documentation and processes meet regulatory standards.
- Document Decisions and Rationale
- Document decisions related to regulatory compliance, including rationale for enforcement discretion or alternative compliance strategies based on FDA policies.
- Document compliance with FDA’s conditions for enforcement discretion, if applicable.
- Plan for FDA Submissions
- Assess whether FDA premarket submissions are necessary for your LDTs based on FDA requirements, the tests intended use and classification according to the FD&C Act necessary to reasonably assure safety and effectiveness.
- Develop timelines and strategies for compiling necessary documentation and data to support submissions, if applicable.
How Beaufort Can Help
The phased approach aims to enhance FDA oversight while allowing laboratories time to adapt to new regulatory standards. However, laboratories should proactively prepare for compliance to ensure continued market access for their LDTs.
Our team of regulatory, quality, and clinical experts can help laboratories navigate the new regulatory landscape and ensure readiness for upcoming compliance milestones. We offers comprehensive support including:
- Training
- Develop and conduct staff training sessions to ensure thorough understanding of and preparedness for the new regulatory requirements.
- Customize training materials to address specific impacts of the FDA’s evolving standards on daily operations.
- Incorporate interactive elements and case studies to enhance comprehension and application of compliance measures
- Assessment
- Assess LDT current inventory and pipeline, determine the FDA’s applicable regulations and enforcement discretion policies that apply to each type of LDTs
- Conduct gap assessments to identify necessary modifications in current quality systems, processes, operations, and documentation.
- Address Quality Management System (QMS) gaps integrating both CLIA and FDA requirements
- Develop regulatory strategies inclusive of considerations of NYSDOH CLEP approvals
- Identify test(s) that may require additional analytical or clinical evaluation
- Compliance
- Map compliance timelines
- Document decisions related to regulatory compliance, including rationale for enforcement discretion or alternative compliance strategies based on FDA policies
- Develop new Standard Operating Procedures (SOPs)
- Prepare 21 CFR 820-compliant design control documentation
- In the event premarket notifications are required, facilitate discussions with the agency through the pre-submission process, and prepare necessary pre-market submissions (Q-Sub, 510(k), De Novo, HDE, PMA)
- Readiness
- Conduct mock inspections
Contact us to learn more about our services and how we can assist in meeting FDA requirements and maintaining market access for your LDTs.
Recently there have been several US and OUS IVD-focused new or proposed regulatory requirements with the potential to impact IVD and LDT manufacturers’ quality management systems (QMS). The implications of these evolving pre- and post-market requirements are wide-ranging and require a clear understanding to inform a strategic yet pragmatic approach for implementation. An overview of these recent regulatory requirements and proposals follows:
QSR to ISO 13485:2016
On January 31, 2024, the FDA issued the Quality Management System Regulation (QMSR) Final Rule, amending the device good manufacturing practice (CGMP) requirements of the Quality System (QS) Regulation under 21 CFR 820 to align most closely with the international consensus standard for Quality System Systems, ISO 13485:2016. Substantively, there is no change to the scope of FDA expectations for a comprehensive quality management system.
The FDA refers to the revised 820 as the “Quality Management System Regulation” (QMSR) and uses the terms “Quality System (QS) Regulation” or “QS regulation” when referring to the unrevised 820.
Incorporation by reference[1] of the QMS requirements of ISO 13485:2016 and Clause 3 of ISO 9000:2015, will provides a similar level of assurance in a firm’s quality management system and ability to consistently manufacture devices that are safe and effective and otherwise in compliance with the Federal Food, Drug, and Cosmetic Act (FD&C Act). However, this amendment makes the integration of risk management into quality systems much more explicit, with changes to terminology that aligns with ISO 13485 – such as the elimination of design history file and device history record.
The FDA extended the proposed transition period from one to two years. FDA will enforce the QMSR requirements beginning February 2, 2026. The changes introduced in the amendment will require a comprehensive gap analysis to gain a clearer understanding of how to align your QMS with the new QMSR. This potentially presents a large scope of work for all manufacturers, especially for those that have previously only conducted business domestically and have less experience with the ISO standard.
Our specialized experience can support your efforts to ensure compliance with the following services:
• QMS design and implementation
• Process and documentation review
• Gap assessment & remediation
• SOP development
• Auditing
• Inspection Readiness
• Training
Laboratory Developed Tests (LTDs)
Historically, FDA has generally exercised enforcement discretion over most Laboratory-Developed Tests (LDTs). When the Medical Device Amendments of 1976 were introduced, LDTs were often lower-risk IVDs developed and used in a single laboratory. LDTS were often developed to address an unmet medical need, e.g., targeting rare diseases for which there were no commercial tests available because the tests would be used so infrequently as to be deemed unprofitable by most companies. Today’s LDTs are developed, manufactured, and used more in-line with those cleared or approved by the FDA; they are often complex, instrument-based assays manufactured in high volume and used to test patient samples shipped in from across the United States.
Due to the impact that the use of diagnostics, including LDTs, is having on patient care, as well as concerns about safety and accuracy of LDTs, FDA has proposed phasing out the general enforcement discretion approach they have taken with most types of LDTs, to make explicit that “in vitro diagnostic products” as defined in 21 CFR 809.3 include those devices manufactured by a laboratory.
Manufacturers of certain types of LDTs will need to be prepared for increased FDA scrutiny during the four-year phaseout period and be fully in compliance with requirements for IVDs by the conclusion of the phaseout program.
With the rule under review by the Office of Information and Regulatory Affairs (OIRA) within the Office of Management and Budget (OMB) since early March, the FDA’s goal of finalizing the rule by Q2 of 2024 appears to be within reach.
Our team can assess and support the impact on your new and existing product development with:
• Classification assessments
• QMS design and support
• LDT verification and validation
• FDA Inspection readiness
• Pre- and post-market submission support
Read more details in our blog here: https://beaufortcro.com/blog/fdas-proposed-rule-for-ldts/
IVDR
To mitigate the risk of shortages for certain high-risk IVDs, on January 23, 2024, the European Commission posted a proposal aimed at further extending the transition period for Regulation (EU) 2017/746 (IVDR). As part of the proposal, the transition period for class D devices, those with high individual and public health risks, would extend until December 2027. Class C devices would have a transition period until December 2028, and class B and class A sterile devices would follow, with a transition period ending December 2029. This extension will only apply to CE-marked IVDs that are already on market and comes with additional conditions.
Manufacturers of all classes of IVDs must have IVDR-compliant quality management systems in place by 26 May 2025, which means that class C or class B and class A sterile devices may be working with less time to align their quality management systems than had previously been anticipated. The manufacturer must also submit an application to the notified body indicating the intent to transition to the IVDR in order to qualify for the extended transition. The deadline for these applications is May 2025 for class D devices, May 2026 for class C devices, and May 2027 for class B and class A sterile devices. In a press release dated February 21, 2024, the European Council stated they have endorsed the updates to Regulation (EU) 2017/746; publication in the Official Journal of the EU could occur as early as April 2024.
Beaufort can help translate all of the complexities to understandable requirements and support all aspects of your implementation with the following:
• Legacy and new device support
• Conformity assessments
• IVDR-compliant QMS assessments and implementation
• Technical documentation preparation and review
• Clinical evidence strategies
• Performance Study Documentation preparation
• Responses to Competent Authority / EC Review questions
• Labeling and UDI compliance
• Distance sale (“LDT”) requirements
Read more here: https://beaufortcro.com/blog/the-potential-impact-of-the-eu-commissions-proposal-to-delay-ivdr-implementation/
Learn More About How Beaufort Can Help
At Beaufort, we specialize in guiding manufacturers through the intricacies of the latest regulatory proposals and updates with expertise in global regulatory affairs and quality consulting, clinical trial services, and data sciences.
Read our latest solutions brochure or contact us today for a consultation.
Contact UsResources:
1Incorporation by reference (IBR) allows Federal agencies to comply with the requirement to publish rules in the Federal Register and the Code of Federal Regulations (CFR) by referring to material already published elsewhere. The legal effect of IBR is that the material is treated as if it were published in the Federal Register and CFR. This material, like any other properly issued rule, has the force and effect of law. (https://www.archives.gov/federal-register/write/ibr#:~:text=Incorporation%20by%20reference%20(IBR)%20allows,to%20material%20already%20published%20elsewhere. Accessed March 13, 2024 and https://www.ecfr.gov/incorporation-by-reference Accessed March 13, 2024)

Impact on LDT manufacturers
Following last week’s presentation and group discussion at AMDM’s IVD Focus Meeting, FDA held a webinar on October 31st to review its Proposed Rule regarding Laboratory Developed Tests (LDTs) aimed at helping to ensure safety and effectiveness of LDTs. The Proposed Rule would amend the Code of Federal Regulations to make explicit that in vitro diagnostic products (IVDs) are devices under the Federal Food, Drug, and Cosmetic Act “including when the manufacturer of these products is a laboratory.”
The Proposed Rule describes a phaseout of FDA’s existing general enforcement discretion approach and would bring certain LDTs under the same regulations as other IVDs. FDA’s rationale for greater oversight reflects its view that healthcare decision makers are increasingly relying on diagnostic testing, and the critical importance of reliable and valid test results to protect public health.
The FDA believes the Proposed Rule will “advance responsible innovation by both laboratory and non-laboratory IVD manufacturers alike by better assuring the safety and effectiveness of IVDs offered as LDTs and removing a disincentive for non-laboratory manufacturers to develop novel tests.”
Proposed Rule Timeline
The phaseout of the FDA’s general enforcement discretion is proposed to occur in five stages over four years. This would allow for greater monitoring and assessment of LDTs to protect patients from any potential excessive risk without disrupting the LDT market. At the conclusion of the phaseout program, LDTs would be generally required to follow the same enforcement requirements as IVDs, unless they fall within specific exclusion categories outlined in the Proposed Rule.
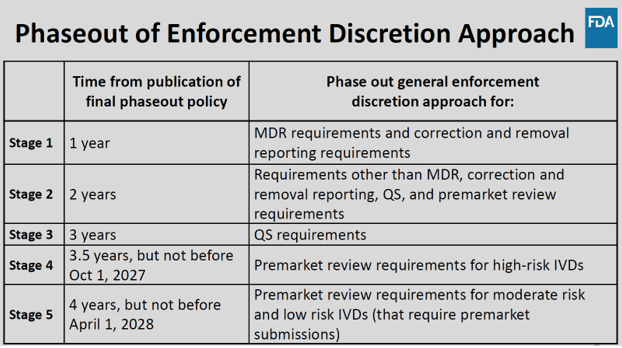
Exclusions and Tests Unaffected by New Rule
The FDA outlines specific categories of tests that are excluded from the Proposed Rule, as “FDA has generally expected applicable requirements to be met,” and include:
- Tests that are intended as blood donor screening or human cells, tissues, and cellular and tissue – based products (HCT/Ps) donor screening tests required for infectious disease testing under 21 CFR 610.40 and 1271.80(c), respectively, or for determination of blood group and Rh factors required under 21 CFR 640.5
- Tests intended for emergencies, potential emergencies, or material threats declared under section 564 of the FD&C Act
- Direct-to-consumer tests intended for consumer use (without meaningful involvement by a licensed healthcare professional)
Categories of tests unaffected by the Phaseout policy include:
- “1976-Type LTDs”
- Human Leukocyte Antigen (HLA) tests
- Forensic Tests
- Public Health Surveillance Tests
Comments to FDA
The Proposed Rule can be accessed on the Federal Register and public questions and comments can be submitted to the FDA until December 4th. A final rule will be released after FDA has read and responded to comments. All LDT regulations FDA describes will become effective 60 days after the date it is published in the Federal Register.
FDA’s presentation can be viewed here:
https://www.fda.gov/media/173457/download?attachment
The transcript will be available here:
https://www.fda.gov/training-and-continuing-education/cdrh-learn
You can view the full Proposed Rule here:
https://www.govinfo.gov/content/pkg/FR-2023-10-03/pdf/2023-21662.pdf
You can submit your comments here:
Federal Register : Medical Devices; Laboratory Developed Tests
How we can help
The Proposed Rule represents a potentially dramatic shift for LDT manufacturers. Beaufort can assess the impact and provide support due to the Proposed Rule for LDTs. Contact us today for an initial consultation.
Keys to managing your successful EUA transition.
On May 11, 2023, the COVID-19 public health emergency (PHE) declared under section 319 of the Public Health Service (PHS) Act expired. However, it is important to note that the Emergency Use Authorization (EUA) declaration under section 564 of the United States (U.S.) Federal Food, Drug & Cosmetic (FD&C) Act is distinct from the PHE and the EUA remains in effect, allowing FDA to use their EUA authority to authorize new tests.
FDA issued two final guidance documents earlier this year on March 27th outlining the next steps manufacturers should take for products granted an EUA during the COVID-19 pandemic. Of the two, FDA’s document entitled “Transition Plan for Medical Devices Issued Emergency Use Authorizations (EUAs) Related to Coronavirus Disease 2019 (COVID-19)” applies to devices, including in vitro diagnostics (IVDs), and provides guidance both for manufacturers that intend to continue distribution after the transition period, as well as manufacturers who do not intend to continue distribution, but who may have some distributed devices still in use.
As it is not yet known when the EUA declaration under section 564 will be terminated, FDA guidance is recommending manufactures begin preparing for the termination date. This includes developing a Transition Implementation Plan, determining the appropriate regulatory pathway, and submitting for market authorization if they wish to continue distribute their product when the EUA declaration ends.
THE 180-DAY WINDOW
As noted, the EUA declaration is distinct from the PHE and currently remains in effect. The FD&C Act requires the Health and Human Services (HHS) Secretary to provide advance notice that an EUA declaration will be terminated (which terminates all EUAs issued under that declaration) and to publish such notice in the Federal Register.
HHS will issue 180-day advance notice of termination of each device EUA declaration either individually or simultaneously. In this transition period, device manufacturers who intend to market and distribute COVID-19 IVDs after the termination of their EUA must prepare and submit traditional regulatory marketing applications (510(k), De Novo, or PMA).
After the 180 days, devices may continue to be labeled and distributed as previously authorized under the EUA provided that the manufacturer has submitted an administratively complete application, excluding pre-submissions, and the application has been accepted by FDA for substantive review.
TRANSITION IMPLEMENTATION PLAN
FDA recommends inclusion of a “Transition Implementation Plan” in the marketing submission cover letter. This plan should address the manufacturer’s intended approach for devices that have already been distributed, (i.e., finished, labeled devices that are no longer in the manufacturer’s possession but are in a 3rd party’s inventory, in distribution in the US supply chain, or in possession of the end user).
In addition to an estimate of the number of devices in distribution, the Transition Implementation Plan should cover the actions that will be taken for either regulatory outcome:
- In the event of a negative decision, the plan should include a disposition plan, or a rationale and subsequent considerations should the manufacturer choose to leave their device in distribution.
- In the event of a positive decision, pathways for stakeholder notification, UDI compliance, registration and listing updates, and labeling updates should be specified.
QUALITY MANAGEMENT CONSIDERATIONS
Non-traditional device manufacturers that may not have a quality system in place that fully complies with 21 CFR Part 820, but who wish to continue to distribute their devices after the EUA termination date, may choose to request an exemption or variance from a device QS requirement as outlined in 21 CFR 820.1(e) and section 520(f)(2) of the FD&C Act within 90 days of publication of the advance notice of termination of the EUA declaration.
CLIA CATEGORIZATION CONSIDERATIONS
IVDs authorized under an EUA are generally authorized for use in specific settings, such as those certified under the Clinical Laboratory Improvement Amendments of 1988 (CLIA) as laboratories, that meet the requirements to perform high or moderate complexity tests, or for use at the point-of-care (POC), i.e., in patient care settings operating under a CLIA Certificate of Waiver, Certificate of Compliance, or Certificate of Accreditation.
- IVDs with an EUA specifying use in laboratories CLIA-certified to perform moderately complex tests will follow the typical categorization process and FDA will categorize the device’s complexity immediately following final action on the submission.
- For IVDs authorized under an EUA for use in CLIA-waived patient care settings, FDA intends to accept submissions under Dual 510(k) and CLIA Waiver or Dual De Novo and CLIA Waiver
- IVDs authorized under an EUA for home use will be categorized as waived without the need for a CLIA Waiver by Application, provided that the marketing submission is cleared, approved, or authorized for home use.
FDA is recommending that any marketing submission that may necessitate a CLIA categorization decision (e.g., tests intended for use in moderate complexity laboratories or in CLIA Certificate of Waiver settings) be submitted as soon as possible to facilitate FDA’s review of the marketing submission and CLIA categorization request (or CLIA Waiver by Application) prior to the termination of the EUA declaration to reduce the potential for disruption in distribution and use.
FOR MANUFACTURERS DISCONTINUING PRODUCT MARKETING
FDA also encourages manufacturers who do not intend to keep their devices on the market to follow a Transition Implementation Plan and to work closely with device distributers, healthcare facilities, healthcare providers, patients, and consumers to assist in transition planning and prevent supply chain disruptions.
If a manufacturer does not intend to continue to distribute its IVD beyond the EUA termination date, the disposition and use of already-distributed devices will continue to be allowed within the product expiry listed at the time of termination and in a manner consistent with the EUA that was in effect. All applicable legal requirements such as adverse event reporting must be upheld. Manufacturers may also choose to voluntarily withdraw their devices from the market prior to EUA termination.
NEXT STEPS
FDA is encouraging manufacturers planning to pursue 510(k) or De Novo pre-market submission authorization for their tests to initiate discussions with the Agency through the Q-submission pre-submission process and to begin working on their marketing submissions, including their Transition Implementation Plans, as soon as possible and prior to the 180-day advance notice of termination.
Marketing submissions must be submitted to and accepted by the FDA before the EUA termination date; on the date of the EUA termination, all EUAs issued under that declaration will be terminated. The enforcement policy in the EUA transition guidance will be in place only for devices that have a marketing submission under review by FDA.
It is important to note that there may be an overflow of marketing submissions as we move closer to the end of the estimated 180-day period, and manufacturers are well advised to submit applications as early as possible to help expedite a prompt decision from FDA.
HOW BEAUFORT CAN HELP
Beaufort’s team of regulatory, quality and clinical experts can provide comprehensive regulatory strategy as well as submission and clinical trial support to afford a successful 510(k), De Novo or Dual CLIA wavier pre-market submission.
Whether for OTC, POC, or laboratory use, we have supported manufacturers throughout the pandemic with their COVID-19 diagnostic test EUAs. We are now helping clients strategize, draft submissions and collect data to expedite their FDA market authorization — and can put this same experience to work for you.