

Impact on LDT manufacturers

Following last week’s presentation and group discussion at AMDM’s IVD Focus Meeting, FDA held a webinar on October 31st to review its Proposed Rule regarding Laboratory Developed Tests (LDTs) aimed at helping to ensure safety and effectiveness of LDTs. The Proposed Rule would amend the Code of Federal Regulations to make explicit that in vitro diagnostic products (IVDs) are devices under the Federal Food, Drug, and Cosmetic Act “including when the manufacturer of these products is a laboratory.”
The Proposed Rule describes a phaseout of FDA’s existing general enforcement discretion approach and would bring certain LDTs under the same regulations as other IVDs. FDA’s rationale for greater oversight reflects its view that healthcare decision makers are increasingly relying on diagnostic testing, and the critical importance of reliable and valid test results to protect public health.
The FDA believes the Proposed Rule will “advance responsible innovation by both laboratory and non-laboratory IVD manufacturers alike by better assuring the safety and effectiveness of IVDs offered as LDTs and removing a disincentive for non-laboratory manufacturers to develop novel tests.”
Proposed Rule Timeline
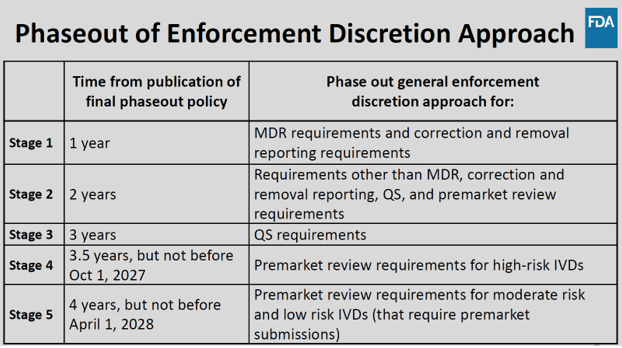
The phaseout of the FDA’s general enforcement discretion is proposed to occur in five stages over four years. This would allow for greater monitoring and assessment of LDTs to protect patients from any potential excessive risk without disrupting the LDT market. At the conclusion of the phaseout program, LDTs would be generally required to follow the same enforcement requirements as IVDs, unless they fall within specific exclusion categories outlined in the Proposed Rule.
Exclusions and Tests Unaffected by New Rule
The FDA outlines specific categories of tests that are excluded from the Proposed Rule, as “FDA has generally expected applicable requirements to be met,” and include:
- Tests that are intended as blood donor screening or human cells, tissues, and cellular and tissue – based products (HCT/Ps) donor screening tests required for infectious disease testing under 21 CFR 610.40 and 1271.80(c), respectively, or for determination of blood group and Rh factors required under 21 CFR 640.5
- Tests intended for emergencies, potential emergencies, or material threats declared under section 564 of the FD&C Act
- Direct-to-consumer tests intended for consumer use (without meaningful involvement by a licensed healthcare professional)
Categories of tests unaffected by the Phaseout policy include:
- “1976-Type LTDs”
- Human Leukocyte Antigen (HLA) tests
- Forensic Tests
- Public Health Surveillance Tests
Comments to FDA
The Proposed Rule can be accessed on the Federal Register and public questions and comments can be submitted to the FDA until December 4th. A final rule will be released after FDA has read and responded to comments. All LDT regulations FDA describes will become effective 60 days after the date it is published in the Federal Register.
FDA’s presentation can be viewed here:
https://www.fda.gov/media/173457/download?attachment
The transcript will be available here:
https://www.fda.gov/training-and-continuing-education/cdrh-learn
You can view the full Proposed Rule here:
https://www.govinfo.gov/content/pkg/FR-2023-10-03/pdf/2023-21662.pdf
You can submit your comments here:
Federal Register : Medical Devices; Laboratory Developed Tests
How we can help
The Proposed Rule represents a potentially dramatic shift for LDT manufacturers. Beaufort can assess the impact and provide support due to the Proposed Rule for LDTs. Contact us today for an initial consultation.