
6 Strategies to Help You Meet FDA Requirements and Maintain Market Access

On April 29, 2024, the US Food and Drug Administration (FDA) issued a long-anticipated final rule asserting its authority to regulate laboratory-developed tests (LDTs) as medical devices (“Medical Devices; Laboratory Developed Tests” (89 FR 37286) (“LDT Final Rule”)). This rule, effective May 6, 2024, marks a significant shift from the FDA’s previous enforcement discretion for LDTs, impacting stakeholders across the diagnostics industry.
Below are key highlights of the final rule and how laboratories offering LDTs may begin preparing to meet the new requirements and timelines.
Redefining LDTs as In Vitro Diagnostic (IVD) Medical Devices
The FDA’s final rule makes explicit that in vitro diagnostic products are medical devices under the Federal Food, Drug, and Cosmetic Act (FD&C Act; 21 CFR § 809.3) “including when the manufacturer of the IVD is a laboratory”. While the laboratory community views LDTs as laboratory testing services, FDA defines LDTs as IVDs that are intended for clinical use and that are designed, manufactured, and used within a single laboratory that is certified under the CLIA and meets the regulatory requirements under CLIA to perform high complexity testing.
Phaseout of Enforcement Discretion
Over the next four years, the FDA will phase out its enforcement discretion for most LDTs. By May 6, 2028, all IVDs offered as LDTs must comply with FDA regulations akin to those for other FDA-regulated medical devices. This includes implementing robust quality management systems, adhering to rigorous reporting practices, fulfilling establishment registration and device listing obligations, and undergoing FDA premarket review where applicable. The phased approach aims to provide laboratories with time to adjust to the new regulatory landscape while ensuring patient safety and maintaining access to accurate diagnostic testing.
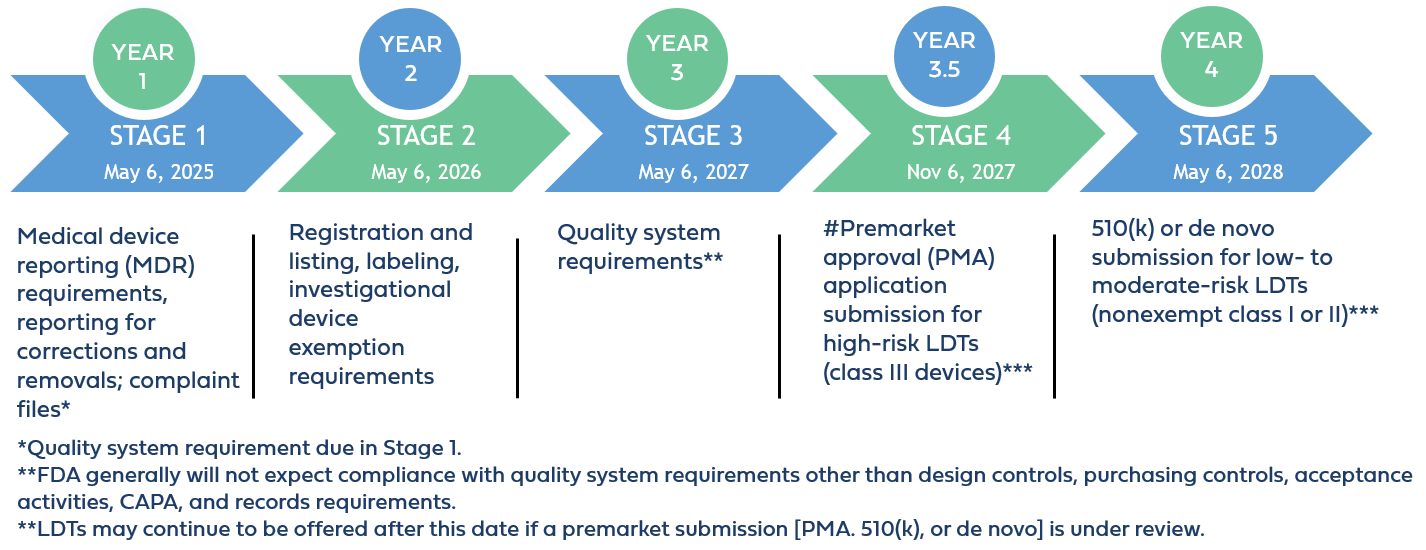
The phaseout policy applies to “IVDs offered as LDTs” defined by the FDA as IVDs that are manufactured and offered as LDTs by laboratories that are certified to perform high complexity testing under CLIA, and used within such laboratories, even if those IVDs do not fall within FDA’s traditional understanding of an LDT because they are not designed, manufactured, and used within a single laboratory.
Implementation Stages and Requirements
STAGE 1: Medical Device Reporting Requirements
Beginning on May 6, 2025, which is 1 year after the publication date of the final LDT rule, laboratories offering IVDs as LDTs must comply with Stage 1 requirements, which include medical device reporting requirements (21 CFR part 803), corrections and removals (21 CFR part 806), and quality system (QS) requirements for establishing and maintaining complaint files (21 CFR 820.198).
These requirements will be new to laboratories because they are distinct from those under Clinical Laboratory Improvement Amendments (CLIA) (42 CFR Part 493). While CLIA mandates procedures for managing complaints related to the quality of laboratory testing services, the FDA’s requirements under 21 CFR 820.198 specifically pertain to managing complaints related to medical devices. This includes establishing and maintaining procedures to receive, review, and evaluate complaints alleging device deficiencies.
STAGE 2: Registration and Listing, Labeling, and Investigational Device Exemption Requirements
Beginning on May 6, 2026, laboratories offering IVDs as LDTs must comply with Stage 2 requirements. These encompass establishment registration and device listing obligations (510 of the FD&C Act, 21 CFR part 607, and 21 CFR part 807 (excluding subpart E)), labeling requirements (section 502 of the FD&C Act and 21 CFR parts 801 and 809, subpart B); and investigational device exemption requirements (section 520(g) of the FD&C Act and 21 CFR part 812).
Compliance will demand understanding and adherence to FDA specifications for labeling, including formatting and content stipulations under 21 CFR parts 801 and 809, as well as ensuring the inclusion of Unique Device Identifiers (UDIs) on labels for traceability throughout the device lifecycle. Establishing a relationship with a UDI issuing agency is necessary to obtain UDIs and assign them to each device model or version, ensuring that the identifier contains essential information such as device identifier, production identifier, and, where applicable, lot or serial number.
Laboratories must also adhere to FDA’s regulatory framework for investigational use of medical devices, when IVDs offered as LDTs are used in clinical investigations. This will include determining the appropriate IVD risk (significant, non-significant, or IDE exempt), obtaining necessary study approvals, and ensuring ongoing compliance throughout the investigation.
STAGE 3: Quality System Regulations (QSR) Implementation
Beginning on May 6, 2027, laboratories offering IVDs as LDTs must comply with Stage 3 requirements, which encompass specific QSR requirements outlined in 21 CFR 820. These include implementing design controls, purchasing controls, and corrective and preventive actions (CAPA).
On February 2, 2024, FDA issued a final rule amending the device QS regulation, 21 CFR part 820, to align more closely with international consensus standards for devices (89 FR 7496, available at https://www.federalregister.gov/d/2024-01709).When the final rule takes effect, FDA will also update the references to provisions in 21 CFR part 820 in this guidance to be consistent with that rule.
Key Components of Design Controls: Central to QSR compliance are Design Controls, a structured approach to product development that ensures devices meet specified requirements and are safe and effective for their intended use. Laboratories developing IVDs as LDTs must establish and maintain processes and procedures that encompass the following key components:
- Design and Development Planning (DDP): Laboratories will be required to create a comprehensive DDP outlining the design control activities and milestones for each device. This plan defines roles and responsibilities of team members involved in the design process and sets out a clear roadmap for development.
- Design Inputs: Laboratories will be required to translate user needs and intended use into specific design inputs. Design inputs encompass functional, performance, and safety requirements that the device must meet to fulfill its intended purpose. These inputs serve as the foundation for subsequent design activities.
- Design Outputs: Laboratories must generate detailed design outputs that specify device characteristics, including drawings, specifications, and other relevant documentation. Design outputs must demonstrate the design meets the design inputs and must be meticulously documented to ensure traceability and compliance.
- Design Reviews: Laboratories must conduct systematic design reviews at critical stages of development to evaluate progress, identify any issues or shortcomings, and verify alignment with design requirements. These reviews ensure that potential problems are identified early and addressed promptly to maintain project timelines and product quality.
- Design Verification: Verification involves rigorous testing and analysis to confirm that the device functions as intended. This process ensures that design outputs meet the specified design inputs and are validated through objective evidence. Verification activities must be well-documented and include testing protocols, results, and conclusions.
- Design Validation: Validation is conducted under defined operating conditions to ensure that the final device meets user needs and intended use. This stage involves comprehensive testing in simulated or actual use environments, including software validation and risk analysis where applicable. Validation activities provide assurance that the device is safe, effective, and suitable for its intended clinical application.
- Risk Management: Throughout the design process, laboratories must implement a systematic approach to identify, analyze, control, and monitor risks associated with the device throughout the design process in compliance with the current version of International Organization for Standardization (ISO) 14971.
- Design Transfer: Ensures a smooth transition of the approved design to production through design transfer activities. This stage includes verification that production processes can consistently produce devices that meet design specifications.
- Design Changes: Laboratories must establish procedures for managing design changes. throughout the device lifecycle. Evaluation, documentation, and validation of design changes are required before implementation to prevent unintended alterations that could compromise device safety, efficacy, or intended use. Any modifications must be carefully controlled and documented to maintain regulatory compliance.
STAGE 4: Premarket Review for High Risk IVDs Offered as LDTs
Beginning on November 6, 2027, laboratories offering Class III IVDs as LDTs must comply with Stage 4 requirements, requiring FDA premarket approval (PMA) before marketing. This entails comprehensive analytical and clinical evaluations to demonstrate device safety and effectiveness, aligning with stringent FDA review standards under 21 CFR 814.20. The PMA application must include all elements required under 21 CFR 814.20, unless omissions can be justified by the applicant.
Separately, the FDA’s Center for Devices and Radiological Health (CDRH) intends to initiate the reclassification process for certain IVDs currently classified as Class III (high-risk devices) to Class II (moderate-risk devices). This reclassification would potentially enable these tests to undergo review through the less burdensome premarket notification (510(k)) pathway instead of the premarket approval (PMA) pathway, the most stringent type of FDA medical device review.
However, the FDA will only consider reclassification for Class III devices if the agency has sufficient information (i.e., experience) to establish special controls that, together with general controls, provide a reasonable assurance of safety and effectiveness for these tests. As a result, only those Class III IVDs for which the FDA can establish appropriate special controls will be eligible for down classification to Class II.
STAGE 5: Premarket Review for Moderate and Low Risk IVDs Offered as LDTs
Beginning on May 6, 2028, laboratories offering Class II moderate-risk and Class I low-risk IVDs as LDTs must submit a 510(k) premarket notification demonstrating substantial equivalence to legally marketed devices, unless exempt. This stage also allows for alternative pathways like De Novo classification requests for devices lacking predicate devices but deemed safe and effective under general or special controls.
Additionally, certain other premarket submissions, such as Humanitarian Device Exemption applications or Biologics License Applications (BLAs), may also be appropriate depending on the specific circumstances of the IVD offered as an LDT.
Of note, for IVDs that rely on or otherwise incorporate software (including off-the-shelf software) or that meet the definition of a “cyber device” (Section 524B(c) of the FD&C Act), premarket submissions will need to be inclusive of documentation for FDA’s review and evaluation. For IVDs offered as LDTs already on the market, if a premarket submission has been received by the beginning of Stage 4 or Stage 5 (as applicable), the FDA intends to exercise enforcement discretion during the review period to ensure continued patient access to these devices without interruption.
The agency’s phaseout transition aims to enhance FDA oversight and ensure the safety and effectiveness of LDTs, by aligning expectations for regulatory compliance with that of commercially distributed medical devices. This phased approach is intended to allow laboratories time to comply with the new regulatory framework while continuing to meet the needs of patients and healthcare providers for accurate diagnostic testing.
Notably the amended regulation and phaseout policy do not change other requirements for laboratories, including requirements under the Clinical Laboratory Improvement Amendments of 1988 (CLIA), which are separate from requirements under the FD&C Act.
Continued General Enforcement Discretion Approach for Certain IVDs
The FDA’s LDT Final Rule, in comparison to its proposed rule, broadens the scope of LDTs for which the agency intends to continue exercising general enforcement discretion. This means that the FDA will generally not enforce any applicable requirements for the following:
- 1976-Type LDTs: These tests use manual techniques and are performed by laboratory personnel with specialized expertise and rely on clinical-use (not research use only) components.
- Certain Human Leukocyte Antigen (HLA) Tests for Transplantation: Specifically, histocompatibility testing when used in connection with organ, stem cell, and tissue transplantation to perform HLA allele typing, for HLA antibody screening and monitoring, or for conducting real and “virtual” HLA crossmatch tests.
- Forensic Tests: Tests intended solely for forensic (law enforcement) purposes.
- US Department of Defense (DoD) or the Veterans Health Affairs (VHA) LDTs: LDTs manufactured and performed within the VHA or DoD and used for patients tested and treated within the DoD or VHA.
Tests manufactured and offered for use exclusively for public health surveillance are also not affected by the phaseout policy.
Targeted Enforcement Discretion Approach for Other IVDs
Additionally, the FDA has outlined a targeted enforcement discretion approach for certain other categories of LDTs. Under this approach, the FDA intends not to enforce certain applicable requirements for these IVDs. This decision is based on the FDA’s assessment that tests in these categories are unlikely to pose significant risks, or they are conducted under circumstances that inherently mitigate potential risks. These IVDs offered as LDTs include:
- Certain Modified Versions of Another Manufacturer’s 510(k) Cleared or De Novo Authorized Test
- LDTs Approved by New York State Department of Health’s Clinical Laboratory Evaluation Program (NYSDOH CLEP)
- LDTs for Unmet Needs Manufactured and Performed by Laboratories in an Integrated Healthcare System
- Currently Marketed LDTs (i.e., prior to May 6, 2024) and “Not Significantly Modified”
- Non-Molecular Antisera LDTs for Rare Red Blood Cell (RBC) Antigens for Transfusion Compatibility
The table below provides a high-level summary of the key categories of LDTs under FDA enforcement discretion and the regulatory requirements to which compliance is expected in Stages 1 through 5 of the agency’s phaseout policy.
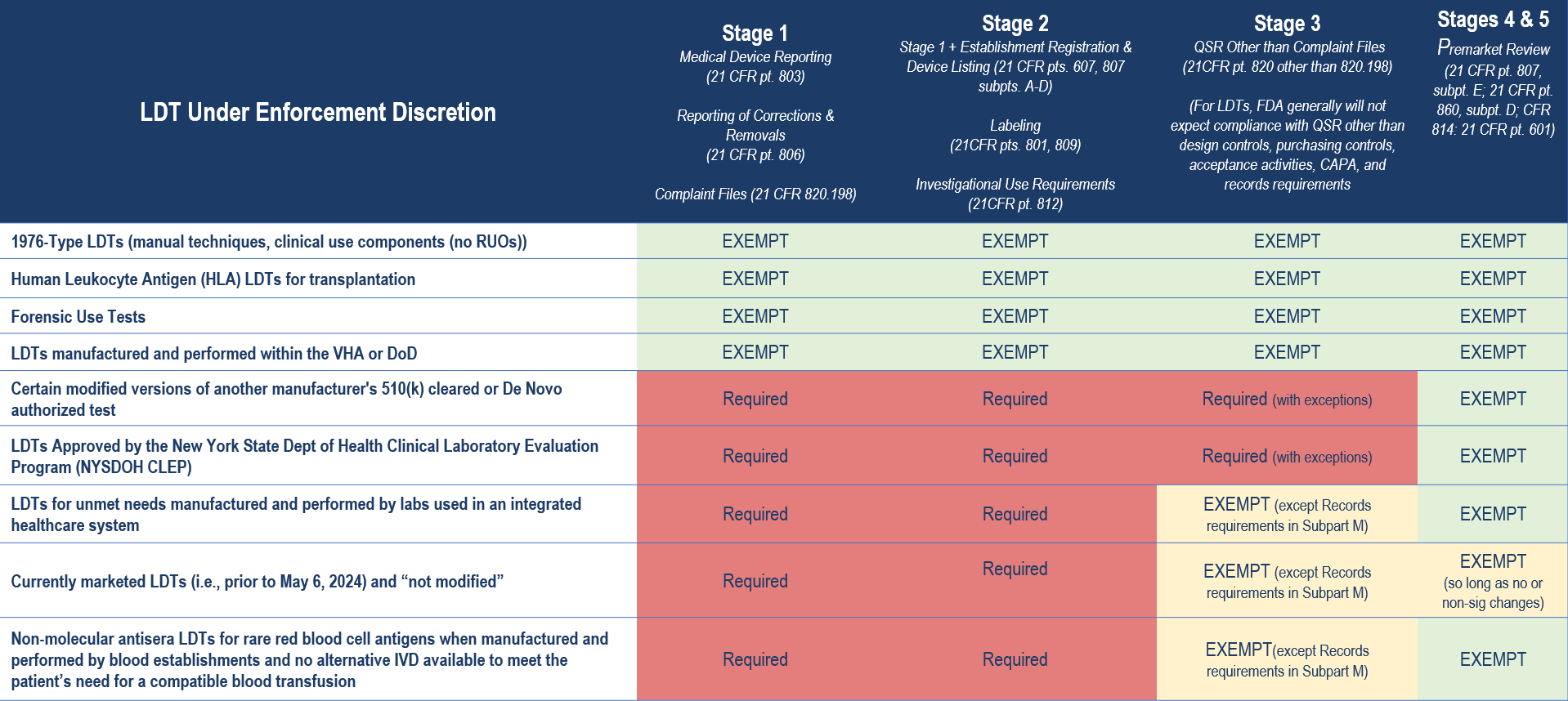
Importantly, there are additional limitations to the agency’s enforcement discretion policies. For example, for LDTs that are modified versions of another manufacturer’s 510(k) cleared or De Novo authorized test, the FDA’s enforcement discretion policy exempting premarket review only applies when the modification was made following design control and other quality system requirements and in a manner that could not “significantly affect the safety or effectiveness of the test and does not constitute a major change or modification in intended use, and where the modified test is performed only in the laboratory making the modification”. Otherwise, FDA expects premarket submissions from laboratories modifying a third party’s 510(k) cleared or De Novo authorized test for the same types of changes for which FDA would expect a premarket submission from the original manufacturer making changes to its own IVD.
Similarly, any “significant modifications” to an LDT marketed before May 6, 2024 will require meeting compliance expectations that align with the phaseout policy. And, for LDTs for unmet needs manufactured and performed by laboratories in an integrated healthcare system, enforcement discretion would end when an FDA-authorized test that addresses the unmet need becomes available.
Moreover, as with any enforcement discretion policy, FDA may update any of these policies as circumstances warrant or if the circumstances that inform these policies change, consistent with FDA’s good guidance practices (section 701(h) of the FD&C Act, 21 CFR 10.115). Additionally, regardless of the phaseout timeline and enforcement discretion policies for certain IVDs, FDA retains discretion to pursue enforcement action for violations of the FD&C Act at any time.
Laboratories will need to determine the FDA’s applicable regulations and enforcement discretion policies that apply to each of their LDTs. This will guide decisions on compliance pathways and documentation requirements and help in determining needed resources.
6 Strategies to Help You Get Started
- Stay Informed
- Stay abreast of legislative developments and regulatory changes that may impact LDT regulation.
- This includes proposed legislation, and changes in enforcement policies as well as FDA announcements, guidance documents, webinars, and other resources.
- Understand FDA Requirements
- Become familiar with FDA definitions, requirements, and expectations for LDTs.
- This includes understanding the differences between LDTs and FDA-cleared/approved tests and knowing when FDA regulation applies.
- Know your Test Status
- Determine the FDA’s applicable regulations and enforcement discretion policies that apply to each type of LDT.
- This will guide decisions on compliance pathways and documentation requirements and help in determining needed resources.
- Quality System Compliance
- Evaluate your laboratory’s CLIA Quality Management System (QMS) against FDA’s Quality System Regulations (QSR) to identify gaps.
- Begin planning necessary changes to align with FDA requirements, ensuring documentation and processes meet regulatory standards.
- Document Decisions and Rationale
- Document decisions related to regulatory compliance, including rationale for enforcement discretion or alternative compliance strategies based on FDA policies.
- Document compliance with FDA’s conditions for enforcement discretion, if applicable.
- Plan for FDA Submissions
- Assess whether FDA premarket submissions are necessary for your LDTs based on FDA requirements, the tests intended use and classification according to the FD&C Act necessary to reasonably assure safety and effectiveness.
- Develop timelines and strategies for compiling necessary documentation and data to support submissions, if applicable.
How Beaufort Can Help
The phased approach aims to enhance FDA oversight while allowing laboratories time to adapt to new regulatory standards. However, laboratories should proactively prepare for compliance to ensure continued market access for their LDTs.
Our team of regulatory, quality, and clinical experts can help laboratories navigate the new regulatory landscape and ensure readiness for upcoming compliance milestones. We offers comprehensive support including:
- Training
- Develop and conduct staff training sessions to ensure thorough understanding of and preparedness for the new regulatory requirements.
- Customize training materials to address specific impacts of the FDA’s evolving standards on daily operations.
- Incorporate interactive elements and case studies to enhance comprehension and application of compliance measures
- Assessment
- Assess LDT current inventory and pipeline, determine the FDA’s applicable regulations and enforcement discretion policies that apply to each type of LDTs
- Conduct gap assessments to identify necessary modifications in current quality systems, processes, operations, and documentation.
- Address Quality Management System (QMS) gaps integrating both CLIA and FDA requirements
- Develop regulatory strategies inclusive of considerations of NYSDOH CLEP approvals
- Identify test(s) that may require additional analytical or clinical evaluation
- Compliance
- Map compliance timelines
- Document decisions related to regulatory compliance, including rationale for enforcement discretion or alternative compliance strategies based on FDA policies
- Develop new Standard Operating Procedures (SOPs)
- Prepare 21 CFR 820-compliant design control documentation
- In the event premarket notifications are required, facilitate discussions with the agency through the pre-submission process, and prepare necessary pre-market submissions (Q-Sub, 510(k), De Novo, HDE, PMA)
- Readiness
- Conduct mock inspections
Contact us to learn more about our services and how we can assist in meeting FDA requirements and maintaining market access for your LDTs.