


SVP Clinical Operations
The Beaufort team was a proud sponsor of The Association of Medical Device Manufacturers (AMDM) Fall IVD Focus Meeting held last week (Oct 19-20) in Los Gatos, CA.
Here are several highlights from the 2023 meeting:
FDA Update On LDT Proposed Rule Changes
On September 29, 20203, the FDA released a Proposed Rule for assuring the Safety & Efficacy of Laboratory Developed Tests (LDTs). This proposal will represent a dramatic shift and phaseout of FDA’s general enforcement discretion approach to LDTs. The AMDM presentation provided an overview of the guidance included the following discussion items:
- Proposed Rule brings LDTs under full FDA regulation and generally fall under the same enforcement approach as other IVDs.
- FDA’s rationale for the Proposed Rule:
- There is no sound basis for regulating LDTs and other IVD tests differently.
- LDT results may not be reliable for patient management use.
- Some LDTs show poor reproducibility of results, lack validation, no clinical data to support use and poor overall performance.
- Evolution and proliferation of LDTs
- Supports increased safety and protection of public health.
- FDA’s rationale for the Proposed Rule:
- FDA was not able to share a timeline but the expected close-out timeline for comments is December 2023.
- Final Rule projected to be published by Spring 2024.
Last week, FDA announced a webinar to be held on Tuesday, October 31st to review the guidance as outlined above as well as answer questions. You can register here:
Software Submission Best Practices
2023 FDA Software Guidance includes a more rigorous look at risk classifications: Risk to the user and patient. For IVDs, software is reviewed as a test system. Tables below, presented by Karen Bijwaard, MS, RAC, MB(ASCP), CQA (CDRH/OPEQ/OHT7/DMGP), show the differences between the 2005 FDA Guidance and the 2023 FDA Guidance language.
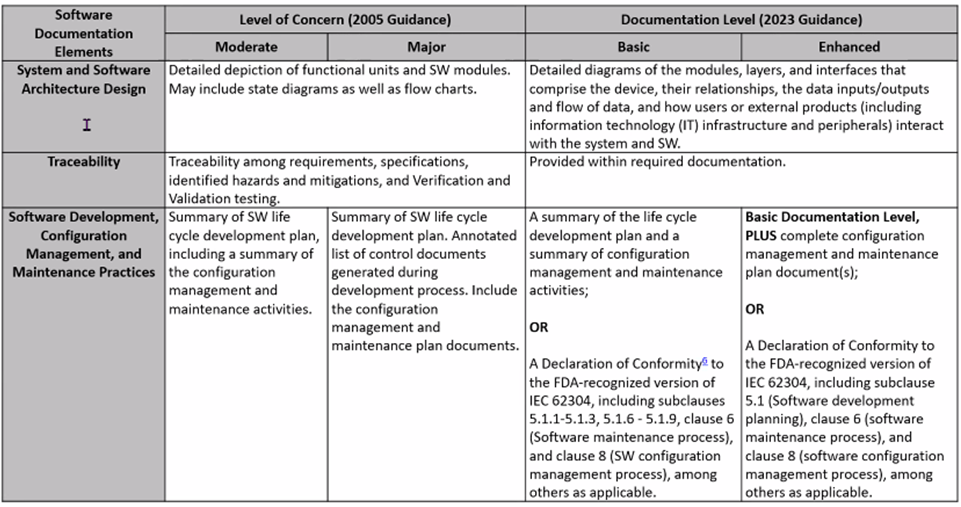
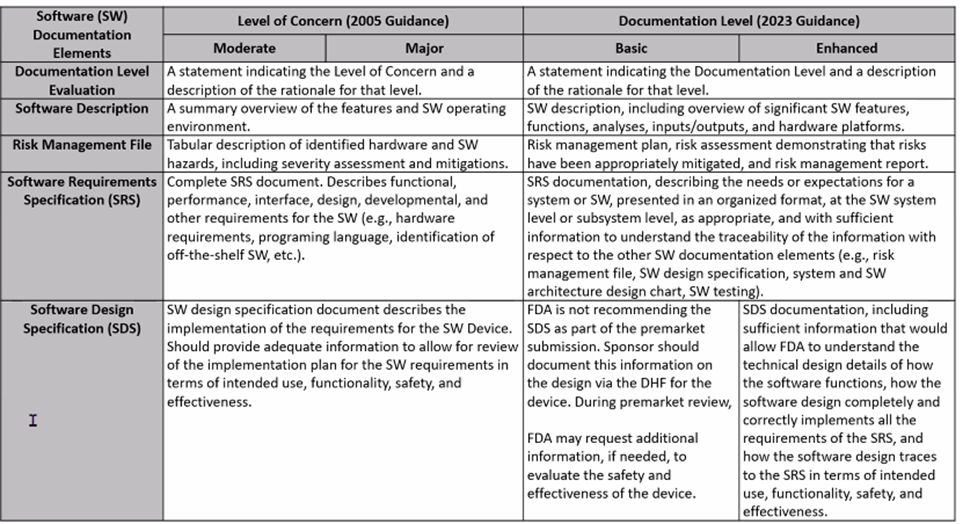
New FDA Voluntary Pilot Program for Oncology Drug Products
FDA announced a voluntary pilot program for Industry with the objective of obtaining/providing greater transparency of minimum performance characteristics that certain tests for certain oncology drugs should meet. This pilot program will not alter the standards for the approval of oncologic drug products or for the marketing authorization of the corresponding companion in vitro diagnostics. FDA Guidance Document was published on 20 Jun 2023.
Additional Meeting Takeaways
- FDA is trying to return to “Normal”
- Reauthorization of Medical Device User Fee Amendments (MDUFA)
- Hundreds of COVID tests under EUA – continue to encourage manufacturers to seek traditional market clearance.
- Review of all submission types has resumed.
- “564” still active – no termination yet.
- Center Initiative Highlights:
- CDRH’s Customer Collaboration Portal: dashboard displays real-time submission status; FDA has received over 21,000 submissions, over 80% of submission come through portal, over 9,000 users.
- eStar: As of October 1, 2023, all 510(k) submissions, unless exempted, must be submitted electronically using eSTAR
- Predetermined Change Control Plans: working on change control protocols – AST is an area where the Agency cleared many protocols.
- Breakthrough Devices Program: >169 designated IVD devices; 18 IVD Devices authorized to market, 6 PMAs approved, 4 510(k)s cleared, 8 De Novos granted.
Contact us today to discuss how any of these issues impact your business – and see how we can help you bring your IVD product successfully through the changing regulatory landscape.