

The AMDM IVD Focus Fall Meeting brought together regulators, industry leaders, and diagnostic professionals for two days of focused discussion and actionable insight. The meeting focused on the current and emerging regulatory landscape for in vitro diagnostics (IVDs), providing clarity on implementation priorities, policy shifts, and the operational realities of compliance in 2026 and beyond.
U.S. Regulation and Policy Landscape
Sessions on U.S. regulatory developments opened with reflections on the federal government shutdown and its operational effects on FDA activities. While essential health and safety functions and reviews supported by existing user-fee funds continue, many new submissions and policy initiatives remain paused until appropriations resume.
A discussion of the Loper Bright legal decision highlighted how the rescinded LDT Rule and evolving interpretations of agency authority will continue to influence FDA’s approach to laboratory-developed tests (LDTs). While FDA’s direct authority over LDTs has been clarified, the Agency may still exert oversight through several indirect pathways — including regulation of collection devices, research-use-only (RUO) products, and requirements tied to companion diagnostics during the associated drug and biologic approvals.
Beyond LDTs, speakers highlighted FDA’s expanding oversight of emerging technologies such as software-based diagnostics and wellness devices. The discussion noted the Agency’s growing attention to wellness wearables — low-risk products that promote healthy lifestyles — and the FDA’s position on software functions that may qualify for general wellness exemptions or fall under stated enforcement discretion.
Across several sessions, a consistent message emerged: even under enforcement discretion, manufacturers are expected to maintain robust design control, validation, and quality management systems.
2025 Legislative Priorities
Meeting discussions highlighted several legislative initiatives likely to shape the next phase of diagnostic policy:
- Protecting Access to Medicare Act (PAMA) reform, aimed at establishing a sustainable reimbursement framework for diagnostic innovation, and the Reforming and Enhancing Sustainable Updates to Laboratory Testing Services (RESULTS) Act. The latter is a proposed bill to reform Medicare’s lab test payment system, address flaws in the current data collection method, and prevent major payment cuts that could threaten patient access to testing.
- The Patients Deserve Price Tags Act, a proposed bill that aims to increase transparency in healthcare costs.
- The Ensuring Patient Access to Critical Breakthrough Products Act, proposed legislation that would create a four-year, automatic temporary Medicare coverage period for FDA-designated breakthrough devices, requiring CMS to make a permanent determination within that time. In contrast, TCET (Transitional Coverage for Emerging Technologies) is an administrative pathway created by CMS, which is more restrictive, limits devices to five per year, requires a separate application, and doesn’t guarantee coverage. Of note, in September 2025, the Ways and Means Committee passed an amended version of the Ensuring Patient Access to Critical Breakthrough Products Act that excludes in vitro diagnostics from scop.
- The Medicare Multi-Cancer Early Detection Screening Coverage Act – proposed legislation to create a pathway for Medicare to cover multi-cancer early detection (MCED) blood tests after they are approved by the FDA and determined to have clinical benefit.
Together, these priorities aim to balance innovation, patient access, and predictable oversight.
Implementing FDA’s Quality Management System Regulation (QMSR)
Although ISO 13485:2016 and the current CGMP requirements for devices in 21 CFR Part 820 are substantially similar, the FDA’s forthcoming Quality Management System Regulation (QMSR), effective February 2, 2026, incorporates ISO 13485:2016 by reference while maintaining key FDA-specific provisions.
Manufacturers already compliant with ISO 13485 should note that several FDA-specific definitions and expectations remain. Beyond the ISO clauses, the QMSR preserves historical Part 820 provisions related to:
- Risk management
- Control of records (including claims, labeling, and packaging)
- Electronic records and signatures compliance
Device manufacturers may need targeted process updates and internal training to ensure full alignment before February 2026.
Global Regulatory Perspectives: IVDR and Health Canada
Transition to the EU In Vitro Diagnostic Regulation (IVDR) remains a critical focus area.
- Class D devices are already transitioned under the IVDR.
- Manufacturers of Class C legacy IVDs must lodge applications to a notified body for conformity assessment by May 26, 2026, and by May 26, 2027 for Class B and A (sterile) devices that were self-declared under the IVDD.
While notified body capacity has improved, review timelines remain long — often 13 to 18 months for combined QMS and product certificates.
Common issues identified during Notified Body reviews include product misclassification, inconsistent intended purpose statements, missing documents required by Annex II/III, and clinical evidence that does not meet IVDR expectations.
Health Canada updates to the Medical Device Regulations (SOR/98-282) focused on recall definitions, reporting and record-keeping, and modernization of MDEL application requirements—changes that took effect December 14, 2024.
Companion Diagnostics (CDx): Coordination and Collaboration
The evolution of companion diagnostics (CDx) continues to highlight the increasing interdependence between drug and diagnostic development.
Conducting early-phase clinical investigations with non-CE-marked products, especially when a CDx may be in scope, remains challenging despite additional MDCG guidance. Key pain points include the adequacy of analytical performance data, clarity on roles and responsibilities (e.g., study sponsorship), and interpretation of clinical evidence when different assays are used within a study or alongside the investigational medicinal product.
EMA consultation timelines and notified body reviews remain gating factors for drug/CDx contemporaneous approval, often driving the need for parallel data strategies across jurisdictions. Successful CDx programs emphasize cross-functional collaboration—linking regulatory, clinical, and analytical teams from concept through commercialization to ensure synchronized regulatory outcomes.
Design and Delivery: Cybersecurity, Self-Collection Devices, and Human Factors Usability Studies
Cybersecurity
Cybersecurity expectations are now embedded across the device lifecycle. FDA’s approach — consistent with NIST SP 800-218 — treats cybersecurity as part of quality system management, encompassing secure design, verification and validation, postmarket monitoring, and coordinated vulnerability disclosure.
Manufacturers were encouraged to document threat modeling, maintain vulnerability management programs, and ensure supplier controls and software updates are traceable to cybersecurity risk mitigations. Documentation should link cybersecurity risk management directly to design controls and product safety — reinforcing that security is intrinsic to device effectiveness, not an afterthought.
Human Factors Usability Studies: Self-Collection and Over-the-Counter (OTC) Devices
Devices intended for use outside professional settings — whether self-collection kits or over-the-counter (OTC) diagnostics — must integrate risk management with user interface design to ensure safety, accuracy, and ease of use.
For self-collection devices, the user interface must be intuitive, logical, and easy to navigate to minimize handling errors and ensure adequate specimen quality. Early formative studies, conducted during development, help refine interface elements and identify use-related risks. Design simplification that prioritizes comfort, clarity, and intuitive interaction improves both sample adequacy and user confidence.
For OTC diagnostics, FDA requires human factors usability studies to demonstrate that untrained users can safely and effectively perform the test and interpret results. Simplified workflows, clear visual cues, and intuitive interfaces improve accuracy and reduce invalid outcomes. Success depends on conducting formative studies early and iteratively, as design changes can introduce new usability issues.
Summative usability studies, performed by intended users in the intended environment and incorporating the complete testing workflow, should confirm that the user interface is free from unacceptable risk.
The key takeaway: whether for self-collection or OTC IVDs, human factors usability studies are integral to safe and effective product performance. Iteration, simplicity, and communication are essential to delivering reliable results and satisfying FDA’s expectations for the growing arena of lay-user diagnostics.
How Beaufort Can Help
As global diagnostic regulation continues to advance, success depends on transforming regulatory complexity into clear, executable strategy. At Beaufort, we help diagnostic and device manufacturers stay ahead of evolving requirements — aligning design, quality, and compliance from the earliest stages of development through market readiness.
Our multidisciplinary team translates policy into practice, guiding manufacturers through FDA and global frameworks with precision and foresight. By integrating regulatory intelligence with operational execution, we enable clients to anticipate change, strengthen quality systems, and accelerate access to innovative diagnostic technologies.
Beaufort helps innovators turn regulatory shifts into market momentum — empowering our clients to advance diagnostic technologies that can make a measurable difference.
Insights from the IVD Track at MedTech Summit 2025
The IVD track at MedTech Summit 2025 in Berlin brought together regulators, notified bodies, and industry experts to examine the continuously evolving regulatory landscape for in vitro diagnostics (IVDs). Over two days, discussions spanned IVDR implementation, the progress of future UK regulation, considerations for IVD and companion diagnostic performance studies, global approaches to in-house and laboratory-developed tests, readiness for EUDAMED, and the first operational experience with EU Reference Laboratories (EURLs). For regulatory leaders, the sessions underscored not only immediate compliance requirements but also broader strategic imperatives:
- Managing portfolios under changing regulations
- Strengthening post-market obligations
- Preparing now for forthcoming demands such as EUDAMED registration and EURL engagement
Success in the IVD space now requires more than regulatory awareness—it demands foresight, adaptability, and a partner who can bridge operational realities with evolving expectations. The conversations in Berlin underscored this need for strategic guidance, not just reactive compliance. Beaufort continues to lead in this space—helping sponsors anticipate change, interpret evolving requirements, and implement pragmatic solutions. The insights below highlight the most critical developments and their implications for diagnostic developers and sponsors.
IVDR Implementation in the EU
Implementation of the IVDR continues to advance. By late 2024, approximately 1,273 (QMS and product) certificates had been issued, including 377 for Class D devices and 32 for companion diagnostics, against an estimated 40,000 IVDs on the EU market.
Navigating shifting regulations requires foresight, not just compliance. Beaufort helps sponsors and manufacturers anticipate evolving requirements, implement pragmatic solutions, and accelerate patient access.
Timelines remain long. Total average time to complete QMS or Technical Documentation certifications each typically require 13–18 months, with much of that time consumed by iterative Notified Body questions and requests for supplementary evidence. Industry and Notified Bodies have submitted proposals to establish predictable timelines.
Transitional provisions. Regulation (EU) 2024/1860 published in July 2024 allows eligible legacy IVD devices to remain available provided they undergo no “significant change” (per MDCG 2022-6), continue to comply with IVDD requirements:

Guidance and interpretation. Although more than 120 MDCG documents are now in circulation, key guidance is still missing and guidance is changing over time (e.g., Covid test down-classification from D to B and changing EMDN numbers) which is complicating planning. Updates to documents such as MDCG 2019-6 on structured dialogue are designed to smooth Notified Body–manufacturer interactions, however the guidance focuses on the “what” without providing the “how”. Additional new guidance is expected for IVDR orphan devices; MDCG 2019-13 on sampling devices for assessment of Technical Documentation; Q&A on the AI Act, PSUR evaluation reports, Q&A regarding IVD performance studies (now available), and a Q&A regarding distance sales among others.
Notified Body Capacity has improved. Notified bodies stressed that staffing investments have eased the bottlenecks. The real challenge now is manufacturers delaying submissions and the quality of manufacturer submissions. Files that are incomplete or inconsistent trigger lengthy review cycles.
The UK MHRA’s Developing Framework
Dual system remains in place. Great Britain (England, Scotland, and Wales) operates under MHRA’s sovereign model, while Northern Ireland continues to align with IVDR. Because the UK’s Medical Devices Regulations 2002 can only be amended rather than rewritten, MHRA has relied on a series of legally required public consultations to move reform forward.
Post-market surveillance is now fully in effect. As of June 2025, PMS obligations apply in Great Britain. These mirror EU IVDR concepts in many respects—risk-based planning, PMS plans and reports, periodic safety update reports (PSURs), trend reporting, and vigilance requirements including incident and FSCA reporting. However, reporting timelines differ, and further MHRA guidance is expected.
Premarket classification will shift to four risk classes. Future statutory instruments will align IVDs with a four-class risk framework based on patient and public health impact. Requirements for market access will be scaled accordingly. MHRA has specifically sought views on regulatory requirements for Class B IVDs, including software IVDs. Options under consideration include allowing manufacturers to self-assess conformity with the regulations while holding ISO 13485 QMS certification from a UKAS-accredited body.
Reliance and recognition models are being developed. MHRA is evaluating pathways that would allow devices already approved by trusted regulators—such as in Australia, Canada, the EU, or the USA—to access the GB market more quickly. These devices would still need to meet GB-specific requirements, including English labelling, a UK Responsible Person, unique device identification (UDI), and compliance with UK post-market surveillance (PMS) obligations.
Fees are increasing. MHRA continues to raise fees in order to sustain its role as a sovereign regulator. This trend will weigh most heavily on smaller developers.
Reform remains a long-term process. Even with consultations and amendments under way, meaningful structural change will take several years. In the meantime, companies must operate under hybrid arrangements while preparing for further reform.
Biomarker Testing in Clinical Trials
Synchronizing drug and diagnostic development timelines is essential to contemporaneous marketing authorizations. Beaufort brings the regulatory and operational expertise to assure IVD submissions are timely and complete, keeping programs aligned.
Defining medical purpose is critical. When a biomarker assay is used to determine patient inclusion or exclusion, guide therapy, or support monitoring, it is deemed to have a medical purpose under IVDR and requires a clinical performance study (CPS). By contrast, assays used solely for stratification or exploratory endpoints may fall outside IVDR scope. However, where data are intended to support CE marking, the trial must still be conducted to IVDR and ISO 20916 standards.
Obligations extend to early-phase studies. Even Phase I trials may trigger IVDR requirements if assay results influence patient management. This includes the obligation for independent monitoring of testing sites, as outlined in Article 68 and Annex XIV.
Combined studies present operational risks. Trials involving multiple pharmaceutical sponsors or diagnostic partners create challenges around data ownership, confidentiality, and clinical trial/performance study timelines. In practice, separate CTA and PS submissions are often the more reliable approach, reducing risk of delay or dispute.
Repurposing CE-marked diagnostics is not straightforward. Using an existing CE-marked diagnostic in a new therapeutic context frequently requires a new clinical performance study, along with updates to the technical documentation, risk management files, and labeling. Sponsors must anticipate these requirements early to avoid disruption.
Pharma-Diagnostic Collaboration – Combine Studies
Early alignment is essential. Regulatory strategies for diagnostics must be developed in parallel with medicinal product plans. Deferring diagnostic planning risks misalignment and late-stage delays.
Roles and responsibilities must be explicit. Collaboration agreements should clearly define who holds responsibility for regulatory submissions, who engages with regulators, and how decision-making authority is exercised across partners.
Rapid response capacity is expected. Regulators such as FDA and EMA often require answers to queries within days or even hours. Effective partnerships now require coordinated rapid-response mechanisms across pharma and diagnostic teams.
Patient access is directly affected. If companion diagnostics are not available at the time of therapeutic launch, drug approvals may stall, or patient access may be delayed. Synchronizing drug and diagnostic timelines is therefore no longer optional; it is a core requirement of development strategy.
Global Perspectives on LDTs
United States. The FDA’s attempt to regulate LDTs through a phased rule was struck down in 2024. Oversight now rests primarily with CLIA (CMS), except where LDTs are used in clinical trials, in which case FDA retains authority under IDE provisions of 21 CFR 812. Industry noted that FDA has signaled concerns when multiple LDTs are used without demonstrating even “minimal” performance; such situations are likely to trigger review discussions if sponsors later seek regulatory approval. In parallel, New York State’s CLEP program continues to operate independently, creating an additional layer of oversight.
European Union. Under IVDR Article 5(5), LDTs are restricted to in-house use within a healthcare institution, and only if no CE-marked equivalent device exists. Full application of these provisions began in May 2024. Article 6 provides the framework for distance sales, requiring that devices supplied to EU patients—whether purchased online or when EU patient samples are tested outside the Union—must still comply with IVDR requirements. In practice, this closes a potential loophole and ensures equivalent regulatory expectations for devices regardless of distribution route.
United Kingdom. Great Britain continues to rely on legacy IVDD-based provisions. Stakeholders see this as an opportunity to design a pragmatic, risk-proportionate framework, particularly for use in clinical trials, which could potentially become a model for future alignment.
Themes and challenges. Regulatory harmonization across jurisdictions remains unlikely. In the EU, reimbursement structures differ significantly by member state, compounding the regulatory complexity. Definitions of what constitutes a “healthcare institution” also vary, which affects feasibility for sponsors. At a broader level, innovation pathways remain poorly defined, and the pandemic experience underscored that preparedness depends on adaptive and flexible frameworks rather than rigid rules.
Post-Market Surveillance: Distributor Interfaces
A strong message came through on PMS under both MDR and IVDR as the new PMS requirements came into force during the conference.
Surveillance extends beyond vigilance. Vigilance addresses the reporting of serious incidents, whereas PMS requires manufacturers to establish systematic and proactive processes to detect trends, analyze data, and act on findings before they escalate into incidents.
The distributor–manufacturer relationship is central. Effective PMS cannot operate in isolation. Agreements must obligate distributors to channel customer complaints, sales representative observations, and user feedback directly to manufacturers. Contracts should require immediate notification, active participation in surveys, and cooperation in implementing CAPAs, field safety corrective actions (FSCA), or recalls.
Risk of regulator bypass. Under IVDR Article 14(6), competent authorities may approach distributors directly. Without robust contractual controls, there is a risk that distributors provide incomplete or unfavorable information. Agreements should therefore require manufacturer review and approval of responses before communications are made to regulators.
UK and EU expectations differ. The UK requires reports even where uncertainty remains, placing greater emphasis on early reporting, on user feedback, and on data gathered from other jurisdictions. Manufacturers operating in both systems must ensure PMS structures can accommodate these differing expectations.
Intersection with the AI Act. For devices incorporating artificial intelligence, the AI Act introduces obligations for monitoring and logging of performance in the field. This will necessitate even closer coordination between manufacturers and distributors to ensure that data capture and reporting are comprehensive and compliant.
EUDAMED – Phased Implementation and Practical Lessons
Phased implementation under Regulation (EU) 2024/1860.
Phased EUDAMED rollout readiness demands more than data entry—it requires cross-functional alignment and proactive planning. At Beaufort, we translate complexity into clear, actionable steps for timely implementation.
- From January 2026, mandatory use of the Actor Registration, UDI/device registration, Notified Body & Certificates, and Market Surveillance modules will apply to new devices.
- The Vigilance module will follow from Q3 2026, with full functionality expected by Q2 2027.
- Development of the Clinical Investigation/Performance Studies (CI/PS) module will continue into Q3 2026. An audit to assess CI/PS alongside the other five modules will take place once its minimum viable product has been delivered.
Operational challenges identified.
- Documentation remains voluminous and often inconsistent, spanning MDR/IVDR, MDCGs, and implementing acts.
- Unpublished “triggers” and discrepancies between the playground and production environments continue to create avoidable failures.
- Data submission routes carry differing risks: manual entry is impractical, XML upload is limited, and machine-to-machine (M2M) connections—preferred by the Commission—take six months or more to establish. Companies starting late are unlikely to achieve readiness in time.
- EUDAMED functions as a minimum viable product with little built-in validation; compliance must therefore be assured within manufacturers’ own systems.
- Portfolio prioritization is uneven. With many companies delaying registration, late surges near deadlines will overwhelm Commission support capacity.
Lessons emphasized at the Summit.
- Treat EUDAMED as a cross-functional initiative, not a project confined to regulatory teams.
- Align with other data-sharing regimes such as FDA’s GUDID and SwissDAMED to reduce duplication and ensure consistency.
- Register devices most likely to generate vigilance reports first to minimize downstream disruption.
- Do not assume the regulation is static; the Commission continues to introduce new and revised requirements with little advance notice.
EU Reference Laboratories (EURLs) – Implementation of Regulation (EU) 2023/2713
IVD Class D conformity assessment now formally involves EURLs
Role and mandated tasks. For Class D devices, EURLs are now formally embedded in the conformity assessment process. Their responsibilities include verifying manufacturer performance claims through independent laboratory testing and confirming compliance with relevant common specifications.
Designation status. As of December 2023, five laboratories had been designated, covering hepatitis/retrovirus, HIV, bacterial agents such as Treponema, and respiratory viruses. Blood grouping and parasitology are not yet covered; however, applications for devices in these categories may still be submitted to notified bodies in the absence of an EURL designation.
Operational launch. EURLs became fully operational on 1 October 2024, when the first physical batch testing began. Harmonized contractual frameworks between laboratories and notified bodies were finalized in December 2024.
Timelines for review. EURLs are required to deliver their opinions within 60 days of receiving the necessary materials. Batch testing applies to each defined production lot.
Different application scenarios.
- New applications submitted after 1 October 2024: both performance verification and batch testing are required.
- Applications lodged before 1 October 2024: batch testing is required, but performance verification is deferred until renewal.
- Devices already certified: batch testing applies immediately, with performance verification added at the next renewal.
Practical challenges observed. Manufacturers are already encountering operational hurdles, including the logistics of transporting batches and instruments, preparation of QC documentation, customs delays, and variability in laboratory transparency. Fees also differ across labs, adding complexity to planning.
Future expansion. A second round of designations is under way to cover blood grouping and additional pathogens, with further EURLs expected to come online in the coming years.
Conclusions
Collaboration is central. Structured engagement across regulators, notified bodies, distributors, and pharmaceutical partners is no longer optional. It is the mechanism through which organizations will navigate change and safeguard patient access.
The regulatory environment remains unsettled. The MedTech Summit 2025 IVD track highlighted that Europe’s IVDR implementation, the UK’s developing medical device regulatory framework, and global divergence in LDT oversight continue to evolve, with new obligations such as EUDAMED adding further complexity.
Strategic leadership is required. For senior regulatory professionals, the challenge extends beyond meeting today’s compliance requirements. Success will depend on anticipating regulatory developments, executing with discipline, and embedding foresight into portfolio and development planning.
How Beaufort Can Provide Support & Expertise
At Beaufort, we work alongside diagnostic developers, pharmaceutical sponsors, and laboratories to navigate the evolving regulatory landscape. Our team brings decades of experience in regulatory strategy, clinical performance study execution, and post-market compliance across global frameworks. We understand the practical realities of engaging with regulators, aligning cross-functional partners, and preparing for new obligations.
As the regulatory climate continues to evolve, the value of a partner who adapts to change, streamlines submissions, and provides operational oversight across jurisdictions is clear. Beaufort combines deep regulatory expertise with hands-on operational experience to ensure programs advance with confidence and patients gain timely access to innovative diagnostics.
For more information on how Beaufort can support your regulatory strategy, contact us directly at [email protected] or provide your contact information here.

From MDUFA to EUDAMED—What Regulatory Leaders Need to Know
The 2025 AMDM Annual In Vitro Diagnostics (IVD) Regulatory Meeting delivered two full days of deep insight, lively discussion, and policy-shaping perspectives across the global diagnostics landscape. Despite the absence of several EU-based presenters due to a travel strike in Belgium and limited engagement from current FDA officials, the sessions offered clarity on pressing regulatory developments and emerging industry priorities.
Day 1: U.S. Landscape, Enforcement, Quality Systems, and Innovation
US FDA Landscape & Policy Direction
- Even with significant disruptions—nearly 3,500 staff impacted by reductions in force and continued institutional knowledge loss through retirements—OHT7 remains committed to meeting MDUFA timelines. However, behind that resilience, growing concerns have emerged around internal restrictions on transparency and open scientific exchange.
QMSR Final Rule
- FDA’s alignment of 21 CFR Part 820 with ISO 13485 through the QMSR final rule is moving ahead as planned, with no indication of delay.
- With a compliance deadline of February 2026, organizations should be actively conducting gap assessments now—particularly around risk management integration, removal of inspection exceptions, training systems, supplier controls, and the management of activities to ensure integrity, inspection, storage, and operations for labeling and packaging.
- Early remediation will position companies for smoother transitions during inspections and audits.- Cybersecurity and Software Requirements.
Software, Cybersecurity & AI/ML IVDs
- Although digital health tools, software as a medical device (SaMDs), and AI/ML-enabled diagnostics are becoming more prevalent, sponsors continue to face challenges meeting the Agency’s expectations. Cybersecurity remains a leading cause of submission refusals and delays with gaps in threat modeling, software bill of materials (SBOM) documentation, and lifecycle security plans persisting.
- Successful development and deployment of AI/ML IVDs rely on implementation of robust design control frameworks and risk management principles, including defined risk-mitigation and post-market monitoring strategies to minimize algorithm bias. Independent data sets for development and test, ideally on study cohort level is one method for minimizing bias and for ensuring reproducible analytical and clinical device performance.
LDT Rule Vacated
- The recent federal court ruling vacating FDA’s final LDT rule has removed near-term compliance obligations but introduced long-term uncertainty.
- While the Court determined regulation of LDTs falls under CMS and CLIA oversight, this is not a return to status quo.
- Questions remain—especially for CDx and single-site tests developed within precision medicine programs. For example, CDER and CBER may still request validation data and rely on CDRH consultation. Early engagement with drug and biologic review divisions is prudent.
- The government may repeal the court decision, but must do so by end of May 2025.
Day 2: Global Regulatory Strategy and Market Opportunities
European Union: IVDR, AI Act, and EUDAMED IVDR Transition
- The phased IVDR transition continues through 2029, but enforcement is already in motion.
- Audits are uncovering deficiencies in compliance with Article 10a—specifically in continuity of supply planning and communication protocols.
- Additional deficiencies should be anticipated if sponsors with legacy devices do not comply with the requirements of Article 10 by the end of May.
EUDAMED Rollout
- Modules for actor registration, UDI, and device listings are set to become mandatory by January 2026. Manufacturers should use the current “playground” environment to test readiness and avoid future data bottlenecks.
AI Act Integration
- High-risk AI systems will face conformity assessments embedded within MDR/IVDR processes. Notified Bodies will need specialized AI evaluation competencies.
New UK Regulations
- While Great Britian’s MDR and EU’s IVDR share many features, their divergence has important implications for global regulatory planning.
- The new post-market surveillance (PMS) regulation (effective June 16, 2025) mirrors many IVDR concepts but adds enhanced expectations around user experience and field safety corrective action notifications. Currently there is no guidance as to whether UKCA-marked companion diagnostics being used in a clinical trial of an investigational medicinal product are expected to apply with PMS requirements due to their UKCA marking.
- The proposed UKCA premarket framework introduces international reliance mechanisms—such as recognition of CE-marked products and approvals from select peer regulators—but not all CDx or SaMDs are expected to be eligible.
- Additional statuary instruments (SIs) are expected in 2025.
Asia-Pacific Region: Acceleration and Modernization
- Across the Asia-Pacific region, significant regulatory modernization is underway
- Japan: Regulatory processes remain rigorous but stable, with CDx demand increasing. Reference pricing reforms are reshaping market access.
- Korea: registration is more difficult than before, however the newly-introduced Immediate Market Entry policy aims to reduce review timelines for cutting edge technologies to under 140 days.. Korea is also developing the first generative AI device review guideline.
- Singapore: released a draft document for IVD medical devices in May 2023, expanding the risk classification to include derivatives of blood, organs, or cell tissues, ensuring to ensure a broader spectrum of IVD devices is adequately regulated.
- China: Continued prioritization of in-country clinical trials, local manufacturing and cost-containment through national volume-based procurement schemes.
- India & Southeast Asia: India’s new classification and adverse event systems aim to harmonize regulatory control.
These developments signal growing regional harmonization with global norms—but each market still requires careful navigation and local expertise.
CDx, Rare Biomarkers, and Sample Scarcity
- Competition for retained sample use among clinical trial enrollment, patient care, scientific research, and CDx development, remains a challenge in precision medicine, particularly in rare disease.
- Real-world data, in silico modeling, contrived samples, and bridging studies are viable when scientifically justified.
- Regulatory flexibility is possible, but only when supported by rigorous rationale and transparent engagement with FDA.
Final Reflections
Across both days, one key message emerged: regulatory harmonization may remain aspirational, but strategic alignment is achievable. The IVD Regulatory Update 2025 reinforced that sponsors must remain agile in response to regulatory change, especially as the FDA, EU, and other regions strengthen expectations around digital health, cybersecurity, and real-world evidence.
Success in 2025 and beyond will depend on a sponsor’s ability to anticipate regulatory expectations, plan with agility, and maintain open, evidence-based communication with health authorities. The IVD Regulatory Update 2025 made it clear that staying ahead requires not only technical excellence but also regulatory intelligence.
To explore how these evolving regulatory developments may affect your organization—and how we can support your IVD product strategy through this period of global change—please get in touch.
Key Changes and Implications for Interventional Clinical Trials
On January 6, 2025, the International Council for Harmonisation (ICH) officially released (adopted) the final version of its Good Clinical Practice (GCP) Guideline E6(R3) (available here: ICH E6(R3) Final Guideline). This updated version reflects the latest standards for the ethical, scientific, and quality conduct of clinical trials involving human participants.
The primary goal of E6(R3) is to protect the rights, safety, and well-being of trial participants, ensuring that clinical trials are conducted ethically and scientifically. The Guideline emphasizes the importance of adhering to principles rooted in the Declaration of Helsinki, while ensuring that clinical trial results are reliable and trustworthy. It applies specifically to interventional clinical trials involving drugs, medicines, medical products, vaccines, and biological products intended for submission to regulatory authorities.
Key Changes in ICH E6(R3)
E6(R3) introduces a significant restructuring compared to previous versions. The main body of the guideline now focuses on general principles, while Annex I outlines the interpretation and implementation of these principles. The revision also retains sections (now in the form of appendices) that provide recommendations for key documents such as the investigator brochure, clinical trial protocol, and protocol amendments. Additionally, a revised glossary is included. A second annex (Annex 2) is under public consultation from November 2024 to March 2025.
Substantial Revisions and New Principles
Among the most notable substantial changes in E6(R3) content are the introduction of two new principles:
- Risk Proportionality: This principle emphasizes that risk management strategies should be tailored to the level of risk posed by the trial, ensuring appropriate oversight and control.
- Roles and Responsibilities: This principle clarifies and expands on the expectations for the various roles in a clinical trial, ensuring accountability and clarity in the responsibilities of all parties involved.
In addition to these new principles, several existing principles have been revised, including those on Ethics, Informed Consent, IRB/IEC Review, Science, and Qualified Individuals. These updates reflect the evolving landscape of clinical trial conduct and regulatory requirements, ensuring that GCP standards remain relevant and effective.
Resources and Training
To help stakeholders understand the new E6(R3) guidelines, the ICH E6(R3) Expert Working Group (EWG) has developed a “Step 4 Introductory Training Presentation,” which provides a high-level overview of the guideline, its development process, and a summary of the key changes. This presentation is available here: E6(R3) Step 4 Presentation.
Additionally, the U.S. Food and Drug Administration (FDA), a founding regulatory member of ICH, plays a crucial role in the development of ICH guidelines, which FDA then adopts and issues as guidance to industry. Following the release of the E6(R3) draft in May 2023 (which we wrote about here), the FDA published a draft guidance titled “E6(R3) Guideline for Good Clinical Practice,” available here: FDA Draft Guidance. While the draft follows the ICH format, the FDA has indicated that the final version will be reformatted to align with its good guidance practices regulations (21 CFR 10.115) before being published.
Conclusion
The adoption of ICH E6(R3) marks a significant step forward in the evolution of clinical trial standards. By incorporating new principles, revising existing ones, and aligning practices with modern trial challenges, this updated guideline promises to improve the quality, safety, and reliability of clinical trials worldwide. As clinical trial professionals, it’s essential to understand changes and integrate them into our daily practices to ensure compliance and uphold the highest ethical standards in trial conduct.
At Beaufort, we share the same vision set forth by the ICH E6(R3) EWG. Their intent to facilitate innovations in clinical trial design and conduct while ensuring participant safety and reliable results is at the heart of what we do.
If you’re looking for a partner that aligns with these principles, come work with us. We are committed to advancing clinical trials with integrity, quality, and an unwavering focus on participant well-being.
Digital solutions can enable an efficient and cost-effective clinical trial while also enhancing participation and compliance.

As the clinical trial landscape evolves, digital technologies are transforming how MedTech (medical device and in vitro diagnostic (IVD)) sponsors conduct clinical research, streamline processes, and achieve better outcomes. In this exclusive Q&A with MPO Magazine, Beaufort’s Karin A. Hughes, Ph.D., Senior Vice President of Global Regulatory & Quality, and Bill Trembley, Senior Vice President and Chief Technology Officer, share their expertise on leveraging innovative digital tools to improve trial efficiency, enhance patient engagement, and meet the unique challenges of today’s MedTech environment. From optimizing workflows to addressing regulatory considerations, their insights provide valuable guidance for sponsors looking to succeed in an increasingly technology-driven field.
As Appeared in MPO Magazine with Sean Fenske, Editor-in-Chief:
In today’s fast-paced MedTech landscape, clinical trials are evolving rapidly spurred by the creation and adoption of innovative digital technologies. For medical devices requiring clinical trials to support regulatory clearance or approval, the stakes are high. Manufacturers want to ensure they are maximizing trial efficiency and are increasingly seeking the best tools to streamline processes and enhance trail outcomes to help them achieve positive results.
Digital technologies can be leveraged to help achieve this goal. There are a number of solutions that are proving invaluable across all trial phases, from improving participant recruitment and informed consent processes to simplifying clinician workflows and enabling better data collection. Similarly, other digital offerings can improve diversity among participants and address barriers faced by underserved communities However, there are considerations to take into account with digital tools, as well.
Speaking to the range of benefits and consideration when using digital technologies to assist with clinical trials is a pair of experts from Beaufort, a CRO specializing in MedTech solutions. In the following Q&A, Beaufort’s Karin Hughes, Ph.D., Senior Vice President of Global Regulatory & Quality, and Bill Trembley, Senior Vice President and Chief Technology Officer, took time to respond to several inquiries on this topic to provide insights on utilizing technology to support sponsors and help clarify any uncertainty.
Sean Fenske: What digital technologies are you reviewing or using in the clinical trials in which you are involved? What advantages do these bring to the trial?
Bill Trembley: The rapid advancement of digital technologies in clinical research offers significant opportunities to improve processes and deliver value for medical device stakeholders. At Beaufort, we are continually evaluating how and where technology can be best leveraged to meet our clients’ unique needs. By streamlining workflows, improving data accuracy, and enabling real-time insights, these innovations drive greater efficiency, reduce administrative burdens, and accelerate timelines, ultimately supporting the success of clinical trials.
Mainstay technologies such as electronic trial master files (eTMF), eSource, and electronic data capture (EDC) systems play a critical role in medical device trials. However, we don’t believe in a one-size-fits-all solution.
Instead, we focus on identifying the right combination of systems tailored to each study’s specific requirements.
This includes seamlessly integrating any legacy systems the sponsor may already use and recommending complementary tools that add value. This approach ensures all technologies work cohesively to achieve goals like improving data accuracy, enabling real-time monitoring, or expediting decision-making.
We know digital technologies now support every stage of the clinical trial process. During the startup phase, site feasibility platforms streamline site activation and participant recruitment, keeping trials on schedule. Tools like eConsent simplify the informed consent process by enabling remote access and fostering greater patient enrollment. Throughout execution, systems like direct data capture minimize manual errors, while automated data cleaning at closeout accelerates database lock and reduces delays. Furthermore, innovations such as wearable devices, remote monitoring platforms, and artificial intelligence enhance the ability to capture real-world evidence and uncover trends that inform better decision-making.
What sets Beaufort apart is our emphasis on integration and customization. By tailoring solutions to each sponsor’s needs and ensuring systems work seamlessly together, we empower our clients to achieve more efficient trials, higher-quality data, and greater confidence in their results.
Fenske: How do patients involved in the trial feel about these technologies? Do they increase participation or compliance?
Trembley: Based on our experience and broad industry data, we know technology can significantly enhance patient engagement and satisfaction. Patients generally feel comfortable with electronic consent technology, which has been shown to improve comprehension compared to traditional paper-based methods. For those who prefer traditional options, we ensure paper-based consent remains available, respecting diverse patient preferences and accessibility needs.
The U.S. Food and Drug Administration (FDA) recognizes that digital tools, when implemented thoughtfully, not only improve engagement but also enhance trial design and conduct. These tools allow for the incorporation of patient input, making trials more patient-centric and increasing enrollment and retention by addressing individual needs and preferences. Digital health technologies (DHTs), such as remote monitoring platforms and wearable devices, further enhance the patient’ experience by enabling data collection from home. This approach reduces the burden of frequent site visits while allowing for continuous monitoring, resulting in more accurate and comprehensive datasets.
As these technologies become more prevalent, addressing privacy and ethical concerns remains a top priority. Tools like eConsent ensure patients are fully informed about how their data will be used and protected, fostering trust in the trial process. Compliance with stringent privacy standards, such as the U.S. Health Insurance Portability and Accountability Act (HIPAA) and the FDA’s regulation regarding Electronic Records; Electronic Signatures (21 CFR Part 11), ensures sensitive health data is safeguarded. Ethical practices, including data usage transparency and patient autonomy, are integral to ensuring patient confidence in these technologies.
We recognize patient-centricity is key to any successful clinical trial. By focusing on end-user experience and addressing privacy and ethical considerations, digital technologies are increasingly seen as enablers of convenience, reduced travel burdens, and a more patient-friendly approach. This growing acceptance leads to higher compliance, better retention, and ultimately more successful trials.
Fenske: What is the reaction from the clinical/physician side? Do they benefit from the incorporation of digital technologies in clinical trials?
Trembley: We see positive technology adoption on the clinician side, especially when the toolsets help reduce administrative burden, streamline workflows, and allow clinical teams to place a greater focus on patient care. For instance, EDC and direct data capture systems simplify data collection and minimize transcription errors, making trial participation less time-consuming for site staff. These efficiencies enable faster data processing and help ensure more accurate and timely reporting of outcomes.
Sponsors benefit significantly as well, with cleaner data, faster timelines, and greater compliance at the site level, leading to more robust and reliable trial results.
However, the key to successful adoption of these technologies lies in comprehensive training and support for site staff. At Beaufort, we recognize familiarity and confidence with new systems are critical to ensuring consistent usage and seamless integration into clinical workflows. Drawing from our experience, we provide tailored training programs that address both the technical aspects of the tools and their practical application within the study’s protocols. This includes hands-on support during startup, ongoing troubleshooting, and clear communication of expectations. By fostering strong partnerships with sites and equipping clinicians with the skills they need, we ensure technology serves as a true enabler, improving trial efficiency and data quality while reducing site burden.
Fenske: Speaking of digital technologies, are you also leveraging these to enhance awareness of a clinical trial? How do they fit into the equation?
Trembley: Absolutely. Digital technologies play a critical role in enhancing awareness and driving recruitment for clinical trials. At Beaufort, we leverage a multi-channel, data-driven approach to effectively target potential participants and increase enrollment rates. For example, we are able to use geographic and behavioral data from sources like the Centers for Disease Control and Prevention (CDC) and National Institutes of Health (NIH), combined with insights from previous studies of similar indications, to identify at-risk populations. This allows us to focus our outreach efforts on those most likely to qualify and benefit from participation.
Our approach can also include the use of digital marketing platforms—social media, search engine marketing, and targeted advertisements—to deliver compelling messaging tailored to specific demographics. Additionally, partnering with patient advocacy groups and community organizations assists in reaching underserved populations, ensuring diversity and inclusivity in our recruitment efforts. This strategy not only enhances awareness but also ensures we meet enrollment goals within established timelines.
Digital tools further complement these efforts by simplifying the enrollment process for participants. By allowing individuals to review and complete consent documents remotely, we reduce barriers to participation and make the process more accessible. These technologies work together seamlessly to enhance trial visibility, attract diverse populations, and support the overall success of the study.
Fenske: Can you explain the relationship between the use of these digital technologies and the FDA’s guidance on Predetermined Change Control Plans for AI-Enabled Device Software?
Dr. Karin Hughes: There is a large spectrum of digital technologies available for potential use in a clinical investigation, including digital technologies for clinical trial process management and digital medical devices for remote data acquisition (including mobile apps).
As Bill described above, digital technologies for clinical trial process management may aid in expediting the clinical trial process by facilitating faster data collection and analysis or by reducing the cost or complexity of clinical trials. These technologies may include those used for clinical trial procedures such as recruitment and enrollment, participant consent, data collection, and data management.
However, depending on the “intended use” of a digital technology, it may meet the definition of a medical device under section 201(h) of the Federal Food, Drug, and Cosmetic (FD&C) Act. When there is a “claim” that a digital technology (e.g., through labeling) is intended to diagnose, treat, cure, or prevent a disease or condition, that digital technology meets the definition of a medical device and is therefore regulated by the FDA. The FDA’s guidance document, “Digital Health Technologies for Remote Data Acquisition in Clinical Investigations” provides recommendations on and considerations for the use of DHTs to acquire data remotely from participants in clinical investigations evaluating medical products. It includes regulatory considerations and guidance on whether engagement with the FDA may be necessary or advisable prior to using such DHTs.
FDA’s guidance document on Predetermined Change Control Plans (PCCPs) for AI-Enabled Device Software applies to FDA-regulated software that meets the definition of a device as outlined in section 201(h)(1) of the FD&C Act. It provides recommendations tailored to artificial intelligence (AI)-enabled medical devices to support iterative improvements through modifications to FDA-regulated AI-enabled devices while maintaining reasonable assurance of device safety and effectiveness. Medical device manufacturers are increasingly leveraging digital technologies, including AI-enabled device software (AI-DSFs), to drive innovation, support healthcare providers, and enhance patient care. Unlike traditional medical devices, which are typically static with fixed design and functionality at the time of FDA review, AI-DSFs follow an iterative development process. These devices are designed to learn and adapt, improving their performance over time.
FDA’s medical device regulatory pathways were initially developed for static devices, requiring supplemental submissions and reviews for any functionality changes. FDA’s guidance on PCCPs for AI-Enabled Device Software addresses the dynamic nature of AI-DSFs by establishing a framework that allows manufacturers to predefine specific modifications to their software while ensuring updates are performed predictably, safely, and while maintaining regulatory compliance. PCCPs are documentation included in a marketing submission for an AI-DSF that pre-specifies what modifications will be made, how they will be assessed (validated and verified), and describes the benefits and risks of implementing the PCCP, along with a plan for risk mitigation. If approved, these plans allow modifications to an AI-DSF over time without necessitating separate submissions and FDA authorizations, potentially accelerating the AI-DSF change process and saving FDA review time.
The PCCP guidance document outlines recommendations on the types of modifications that FDA believes may be appropriate for inclusion in a PCCP for an AI-DSF, as well as the information to include in a PCCP in a marketing submission for an AI-DSF.
Fenske: Generally speaking, what recommendations do you have for medical device clinical trial sponsors that you see as an often overlooked or misunderstood aspect?
Dr. Hughes: When incorporating digital technologies into medical device clinical trials, we recommend sponsors ensure the digital technologies are fit-for-purpose by clearly defining how each technology will be used (e.g., primary endpoint collection, secondary outcomes, or operational support). Sponsors should consider the technical and performance specifications of the digital technology, such as accuracy, reliability, and data output compatibility, and ensure the technology aligns with key features of the study design (e.g., whether the study is centralized or decentralized; the need for real-time data transmission or passive monitoring; etc.).
We also advise sponsors to implement strategies to minimize the introduction of bias or variability and ensure consistency of use across sites and users. This can be achieved through thoughtful planning and specification of usage in their clinical trial standard operating procedures, data management/data transfer plans, risk management plan, and a safety monitoring plan, as well as through robust training and plans for technical assistance to participants or personnel during the trial. It’s equally important for sponsors to ensure their contract research organization has demonstrated expertise in selecting, validating, and operationalizing digital technologies within clinical trials.
Finally, we recommend sponsors ensure they consider applicable regulatory requirements when using digital technologies in medical device clinical trials.
Digital technologies may meet the definition of a medical device. Although such medical devices intended for use in clinical investigations are exempt from most regulatory requirements applicable to devices, the investigation must still comply with applicable requirements under the FDA’s Code of Federal Regulations (CFR) that outline the procedures and requirements for clinical research of medical devices (21 CFR part 812). In submissions to the FDA that rely on digital technologies, sponsors need to be prepared to describe the flow of data from the digital technology to a durable electronic data repository; methods for access control, including revocation of system access; methods for verification and validation to ensure the digital technology is fit for purpose; and mitigations related to data privacy risks, cybersecurity risks, and compliant processes for obtaining informed consent.
Fenske: Do you have any additional comments you’d like to share based on any of the topics we discussed or something you’d like to tell medical device manufacturers?
Dr. Hughes: The integration of digital technologies into medical device clinical trials is no longer optional but essential to advancing the quality, efficiency, and inclusivity of clinical research. While these technologies offer immense potential, medical device sponsors must prioritize thoughtful implementation to unlock their full benefits. This includes ensuring digital tools are fit-for-purpose, validated, and seamlessly integrated into trial workflows to minimize risks and enhance data quality.
Equally important is a patient- and clinician-centric approach to technology deployment. Addressing usability, privacy concerns, and regulatory compliance not only improves adoption but also fosters trust and engagement among trial participants and site personnel. Collaboration with experienced contract research organizations, such as Beaufort, and proactive engagement with regulatory authorities, such as the FDA, can provide critical guidance to ensure trials align with evolving standards and leverage best practices.
As digital health technologies and AI-enabled device software evolve, regulatory frameworks like the FDA’s guidance on PCCPs enable iterative improvements while maintaining safety and compliance. By embracing these frameworks and tailoring technology strategies to each study’s unique requirements, manufacturers can drive innovation, streamline operations, and ultimately improve patient outcomes.
Learn more about our comprehensive Clinical Trial Services & Solutions or Contact Us today.
Navigating the Complexity of the IVDR
With the enactment of Regulation (EU) 2017/746 (IVDR), the landscape for developing companion diagnostics (CDx) has undergone significant transformation. With Eudamed lacking a functional Clinical Investigations and Performance Studies module, sponsors must consider individual national procedures, submitting applications, modification notifications, and final reports to each Member State where CDx studies are conducted.
This regulatory evolution demands that pharmaceutical and diagnostic partners devise new strategies and refine their processes, particularly when conducting a “combined study” that pairs a clinical trial of a medicinal product with a performance study of an in vitro companion diagnostics.
Karin A. Hughes, Ph.D., Beaufort’s SVP Global Regulatory and Quality addressed these pressing issues at the 16th Next Generation Dx Summit. Her presentation, “Clinical Trials with CDx—Navigating the Complexity of the IVDR” provided insights and effective strategies to help sponsors effectively develop a companion diagnostic in the EU.
Dr. Hughes is an industry leader in global regulatory affairs and Beaufort was proud to sponsor this important event to help support the effective development of companion diagnostic products.
Learn more about our comprehensive Companion Diagnostic and IVDR services and solutions or Contact Us today.
Key Considerations to Achieving Contemporaneous Drug and CDx Authorizations
In the realm of precision medicine and targeted therapeutics, a symbiotic relationship between drug and diagnostic co-development is essential. Collaboration across all levels of both organizations, alignment on timelines and goals, and the integration of parallel processes, are all key elements for successful commercialization. While assay development capabilities are important, for pharmaceutical companies, ensuring the regulatory readiness and preparedness of their diagnostic partner is critical and, if not managed effectively, can pose significant financial, regulatory and time to market challenges.
Beaufort offers substantial expertise in assisting pharmaceutical firms throughout the entire co-development process of companion diagnostics (CDx). This encompasses assessing quality assurance measures, such as documenting development processes and assay specifications; designing and developing analytical and clinical performance studies; and supporting consultation with the FDA and other regulators, all essential for ensuring the accuracy and reliability of the companion diagnostic. Our support extends from the pre-clinical stages to clinical trials and market authorization.
Feasibility: Aligning with a Diagnostic Partner
Ideally, pharmaceutical companies are able to identify and partner with a diagnostic sponsor as soon as the potential need for a companion diagnostic (CDx) is recognized. The earlier in the therapeutic product’s development process this partnering occurs, the easier it is to synchronize timelines and identify and address gaps, ensuring that the assay is prepared for clinical trial and that contemporaneous marketing authorizations can be supported.
As drug development and assay development follow different timelines and have unique milestones, design and authorization requirements, it can be burdensome and challenging for pharmaceutical companies to ensure that the CDx partners development processes are robust and aligned to target contemporaneous approvals. Pharmaceutical companies may not be prepared with the knowledge necessary to determine:
- the completeness of the partner’s defined test system: preanalytical components and off-the-shelf instrumentation and whether the design and manufacture of the components comply with applicable requirements under the Quality System regulations, including software and cybersecurity requirements;
- the adequacy of the analytical validity needed to ensure a companion diagnostic is ready for use in the Phase 3 clinical trial;
- the robustness of the partner’s quality assurance processes and manufacturing capabilities to ensure consistent and reliable production of the diagnostic test for Phase 3 studies through commercial use;
- the adaptability of the partner’s quality and regulatory processes in case of changes in project scope or timelines;
- the experience of the partner with pre-clinical, clinical, premarket authorization and post-approval regulatory requirements and the partners capability to support worldwide CDx clinical performance study applications and market authorization submissions;
- the scalability of the partner’s operations to meet increased demand as the pharmaceutical product reaches more markets or indications.
Beaufort recognizes the importance of rigorously evaluating how a company’s therapeutic product development program aligns with the capabilities and processes of a potential or current diagnostic partner – and offers a pragmatic approach to better mitigate risks posed by lack of regulatory readiness.
Central to Beaufort’s approach is the repeated evaluation of a CDx partner’s quality documentation and regulatory compliance preparedness against co-development milestones and timelines. This methodology allows the identification and subsequent closure of gaps, mitigating development risks and ensuring alignment between assay analytical and clinical validation strategies and the therapeutic development milestones of our pharmaceutical partners.
Assay Development: Designing with Therapeutic Milestones in Mind
In certain geographies, including both the United States (US) and the European Union (EU), the regulatory landscape for companion diagnostic (CDx) trials involves several key filings and approvals.
U.S. Considerations
In the U.S., investigational device studies are subject to the Investigational Device Exemption (IDE) regulation (21 CFR 812), even if the therapeutic product(s) in the trial is/are exempt from the requirements of 21 CFR Part 312 under 21 CFR 312.2(b). Before initiating clinical trials involving a companion diagnostic, it may be necessary to obtain an Investigational Device Exemption (IDE) from the Food and Drug Administration (FDA). The need for an IDE is based on the intended use of the CDx in the clinical trial and whether the CDx presents a potential for serious risk to the health, safety, or welfare of a subject. When an IDE is not required, the device study typically must follow the abbreviated requirements at 21 CFR 812.2(b).
Beaufort can support this determination, as well as assist in the development of justification as to why an IDE may not be needed or in the preparation of an IDE application for a significant risk device study. We also support pre-IDE meetings with the FDA to discuss the proposed CDx trial, study design, and regulatory requirements.
EU Considerations
In the EU, devices meeting the definition of an in vitro diagnostic, e.g., CDx, must comply with the requirements of Regulation (EU) 2017/746 (IVDR), including those for performance studies. If an IVD is not CE-marked for its use in a clinical trial, a clinical performance evaluation is required. If, in the clinical trial, the IVD results will be used for medical management decisions, the performance evaluation study is conducted as an interventional study and applications must be submitted for authorization by the Competent Authority (CA) and approval by an Ethics Committee (EC) in each EU Member State where the clinical trial/clinical performance study is to be conducted.
Here, Beaufort has prepared the documentation required to comply with the relevant In Vitro Diagnostic Regulation (IVDR) requirements as well as national application requirements and Ethics Committee guidelines within the individual Member States.
Ensuring CDx Reliability, Reproducibility, and Consistency
Before going to trial the pharmaceutical company should also have confidence that the CDx they’ve chosen can accurately identify the presence or absence of the specific biomarker(s), antibodies or genetic mutations that are crucial for selecting appropriate patients for the drug therapy being developed. The diagnostic should demonstrate reliability, reproducibility, and consistency across different testing conditions and, if necessary, laboratories. These characteristics are essential in ensuring the population identified by the assay during the Phase 3 trial is the same population identified post-approval and during commercial use. Hence, adequate assay analytical verification and stability data along with a demonstration of sufficiently robust “manufacturability” must be available in time for CDx study protocol to undergo review and approval by Institutional Review Boards (IRB)/Ethics Committees (EC). Simultaneously, applications need to be prepared, submitted, and approved by the FDA and/or EU National Competent Authorities, if applicable.
Additionally, understanding the soundness of the available design documentation pre-clinically may aid in minimizing assay changes that can lead to post clinical changes and the need for bridging studies. An effective study design will also ensure the adequacy of available biomarker-positive and –negative samples obtained during trial to provide interpretable clinical study results.
Beaufort’s extensive experience with the quality system and regulatory requirements for IVD development includes analytical protocol and report development and review, ensuring that analytical verification requirements are satisfied. We bring an understanding of how IDE risk determinations, humanitarian use or breakthrough device designations, and IVDR performance study requirements can all impact trial planning.
Aligning Goals for Simultaneous Approvals
Clinical trial protocol development for a therapeutic product trial involving a CDx presents unique challenges, as it requires integrating evaluation of both the diagnostic test and the therapeutic intervention. Often, the CDx will inform the enrollment or management of trial participants. The design of the CDx performance study must guarantee that the data acquired during the trial are sufficient to demonstrate the safety and effectiveness of the CDx for a Class III device PMA in the U.S. and/or provide state-of-the-art clinical evidence for a Class C CDX technical documentation assessment in the EU.
Our team excels in finding that balance and collaborates with pharmaceutical sponsors and their diagnostic partners to design CDx clinical validation strategies that align with the clinical trials design and goals of the therapeutic product.
Beaufort’s approach takes into account factors that add complexity due to the use of a CDx, particularly the differences in documentation requirements that can sometimes delay or impede simultaneous approvals for both the clinical trial and the CDx performance study applications. These differences, if not understood and properly managed, can result in delayed or slowed Phase 3 enrollment, especially in the EU.
Regulatory Submission Planning and Support
A significant challenge faced by pharmaceutical companies is anticipating regulatory readiness issues throughout the process and effectively addressing identified gaps. Prior to regulatory submission, the companion diagnostic must be supported by sufficient data to demonstrate safety and effectiveness of the assay and the assay sponsor must be prepared for Quality Management System (QMS), Bioresearch Monitoring (BIMO), and manufacturing inspections. The chosen submission pathway for the CDx will not only dictate the content of the submission package but also influence the timing of the submission, as review timelines can vary. The documentation required to support a Pre-Market Approval PMA or other diagnostic regulatory submission package is extensive, and pharmaceutical developers may lack access to suitable regulatory and medical writing staff to support the CDx submission.
Beaufort is well-equipped to support the development and submission of U.S. PMA and De Novo Classification requests and EU Technical Documentation as well as conduct mock QMS, IVDR, BIMO and manufacturing audits and inspections to ensure the regulatory readiness of diagnostic partners.
Harmonizing development timelines to achieve contemporaneous marketing approval provides for the fastest time to market. The cost and time ramifications can be substantial when a therapeutic product is ready for regulatory submission, while its companion diagnostic is not. However, alternative strategies are available when contemporaneous approval is not achievable.
Beaufort offers extensive expertise in diagnostic regulatory affairs and CDx co-development, allowing us to tailor an alternative co-development strategy to ensure an assay is commercially available in time for the therapeutic product launch.
How We Can Help
Ensuring regulatory readiness by a companion diagnostic provider is a pivotal aspect of companion diagnostic development, ensuring CDx adherence to quality, safety, and efficacy regulations and standards while achieving contemporaneous marketing approval.
With Beaufort’s extensive expertise within the diagnostic industry and a deep understanding of the complex and evolving CDx global regulatory landscape, we offer our pharmaceutical partners invaluable insights and guidance in evaluating and coordinating with their diagnostic partner. We can conduct comprehensive assessments of their regulatory readiness, identify any gaps and provide resources to address those gaps throughout the co-development process. Our experience spans programs at every stage of development, enabling us to devise regulatory strategies that facilitate the most efficient path to marketing approval.
Contact us today to see how we can help support your current or future diagnostic project.

SVP Clinical Operations
The Beaufort team was a proud sponsor of The Association of Medical Device Manufacturers (AMDM) Fall IVD Focus Meeting held last week (Oct 19-20) in Los Gatos, CA.
Here are several highlights from the 2023 meeting:
FDA Update On LDT Proposed Rule Changes
On September 29, 20203, the FDA released a Proposed Rule for assuring the Safety & Efficacy of Laboratory Developed Tests (LDTs). This proposal will represent a dramatic shift and phaseout of FDA’s general enforcement discretion approach to LDTs. The AMDM presentation provided an overview of the guidance included the following discussion items:
- Proposed Rule brings LDTs under full FDA regulation and generally fall under the same enforcement approach as other IVDs.
- FDA’s rationale for the Proposed Rule:
- There is no sound basis for regulating LDTs and other IVD tests differently.
- LDT results may not be reliable for patient management use.
- Some LDTs show poor reproducibility of results, lack validation, no clinical data to support use and poor overall performance.
- Evolution and proliferation of LDTs
- Supports increased safety and protection of public health.
- FDA’s rationale for the Proposed Rule:
- FDA was not able to share a timeline but the expected close-out timeline for comments is December 2023.
- Final Rule projected to be published by Spring 2024.
Last week, FDA announced a webinar to be held on Tuesday, October 31st to review the guidance as outlined above as well as answer questions. You can register here:
Software Submission Best Practices
2023 FDA Software Guidance includes a more rigorous look at risk classifications: Risk to the user and patient. For IVDs, software is reviewed as a test system. Tables below, presented by Karen Bijwaard, MS, RAC, MB(ASCP), CQA (CDRH/OPEQ/OHT7/DMGP), show the differences between the 2005 FDA Guidance and the 2023 FDA Guidance language.
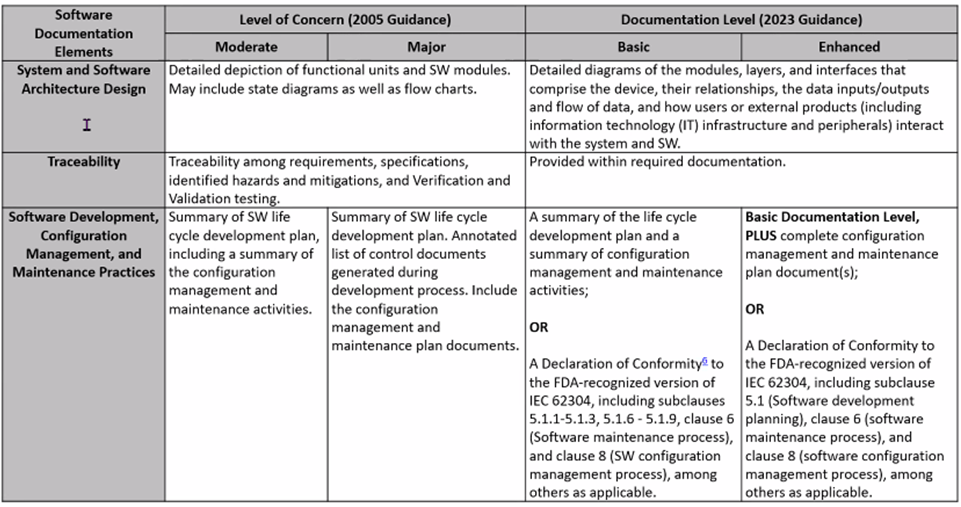
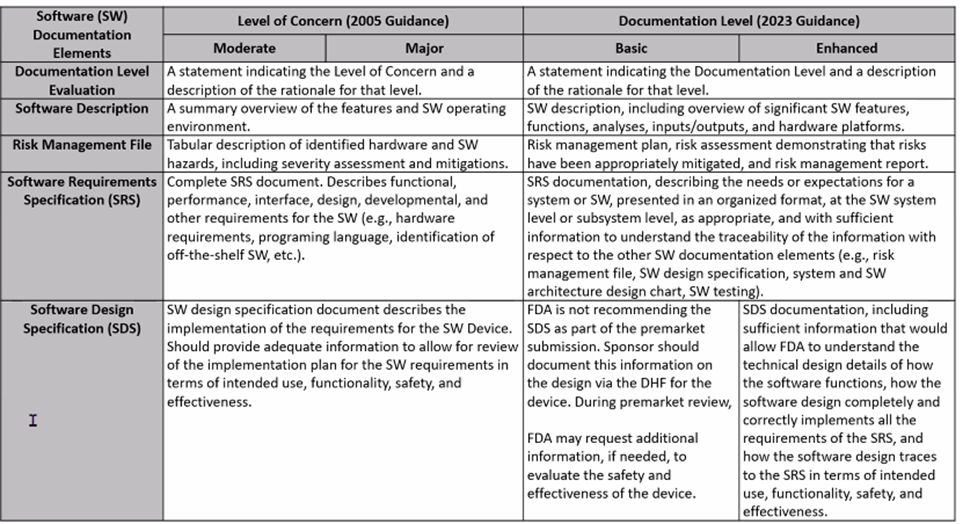
New FDA Voluntary Pilot Program for Oncology Drug Products
FDA announced a voluntary pilot program for Industry with the objective of obtaining/providing greater transparency of minimum performance characteristics that certain tests for certain oncology drugs should meet. This pilot program will not alter the standards for the approval of oncologic drug products or for the marketing authorization of the corresponding companion in vitro diagnostics. FDA Guidance Document was published on 20 Jun 2023.
Additional Meeting Takeaways
- FDA is trying to return to “Normal”
- Reauthorization of Medical Device User Fee Amendments (MDUFA)
- Hundreds of COVID tests under EUA – continue to encourage manufacturers to seek traditional market clearance.
- Review of all submission types has resumed.
- “564” still active – no termination yet.
- Center Initiative Highlights:
- CDRH’s Customer Collaboration Portal: dashboard displays real-time submission status; FDA has received over 21,000 submissions, over 80% of submission come through portal, over 9,000 users.
- eStar: As of October 1, 2023, all 510(k) submissions, unless exempted, must be submitted electronically using eSTAR
- Predetermined Change Control Plans: working on change control protocols – AST is an area where the Agency cleared many protocols.
- Breakthrough Devices Program: >169 designated IVD devices; 18 IVD Devices authorized to market, 6 PMAs approved, 4 510(k)s cleared, 8 De Novos granted.
Contact us today to discuss how any of these issues impact your business – and see how we can help you bring your IVD product successfully through the changing regulatory landscape.