
Updates to the UK Medical Device Regulations

The regulatory landscape for medical devices, including in vitro diagnostics (IVDs), in the United Kingdom (UK) is undergoing a significant transformation. This article outlines key updates to the UK’s future medical device regulations, their implications for the industry, and guidance for regulatory professionals navigating these changes.
Current Regulations Governing the Sale & Supply of In Vitro Diagnostic (IVD) Medical Devices in the UK
The UK’s regulatory framework for medical devices has evolved in response to Brexit-induced divergence from the European Union’s (EU) Medical Device Regulation (MDR) and In Vitro Diagnostic Regulation (IVDR) as well as the need to enhance patient safety while supporting MedTech innovation.
Since January 1, 2021, several changes have impacted how medical devices, including IVDs, are placed on the market in the UK. In Great Britain (GB – England, Wales and Scotland), IVDs are currently regulated under the Medical Devices Regulations 2002 (SI 2002 No 618, as amended) (UK MDR 2002)[1] which remains largely based on the EU Directive 98/79/EC (IVDD)[2].
Under the Northern Ireland Protocol[3], different rules apply allowing Northern Ireland to continue following EU law. Thus, CE marking remains mandatory for devices sold in Northern Ireland, the EU, and the European Economic Area (EEA).
Implementation of the Future Regulations
In 2022, the UK government published its “Government response to consultation on the future regulation of medical devices in the United Kingdom”[1], outlining a phased approach to reform the UK MDR.
In early 2024, the Medicines & Healthcare products Regulatory Agency (MHRA) released a regulatory roadmap detailing the 2024-2025 timelines for delivering the new framework. This roadmap was revised in December of 2024 (version 2.0)[2] to reflect updated timelines.
Key Updates:
- New Post-Market Surveillance (PMS) Regulations – The PMS statutory instrument (SI) was signed into law in December 2024 and will come into force in June 2025 after a six-month transition period.
- Pre-Market SI – Changes will include new IVD classification rules, revised conformity assessment procedures, and new IVD approval pathways including international recognition routes. The necessary SI is expected to be introduced to Parliament in late 2025 and come into force in 2026.
- Software, AI & Digital Mental Health Products – The MHRA is expected to publish new/draft guidance on AI development and deployment, cybersecurity and digital mental health technologies in 2025.
- IVD Policy Development – New policies and guidance for Exceptional Use Authorisation and Early Access and Use are in development, with a dedicated roadmap expected by Q4 of 2025.
NEW Post-Market Surveillance (PMS) Requirements
The Medical Devices (Post-market Surveillance Requirements) (Amendment) (Great Britain) Regulations 2024[6] introduced stricter obligations for manufacturers, aligning UK requirements more closely with the EU IVDR and MDR while maintaining unique GB-specific elements. The MHRA have published guidance[7] to aid in implementation.
Key PMS Changes for IVDs:
- Comprehensive PMS Documentation – Manufacturers must maintain a PMS system, submit detailed PMS plans, and, for certain risk classes, provide Periodic Safety Update Reports (PSURS).
- Stronger Reporting Obligations:
- Serious public health threats – Report within 2 calendar days after the manufacturer becomes aware.
- Death or unanticipated serious deterioration in state of health – Report within 10 calendar days after the manufacturer becomes aware.
- Anticipated serious deterioration in state of health – Report withing 15 calendar days after the manufacturer became aware.
- Field Safety Corrective Actions (FSCAs) – Report immediately upon initiation.
- Field Safety Notices (FSNs) – Distribute to users and stakeholders without delay to mitigate risks.
- Improved Traceability and Transparency – Enhanced supply chain reporting and tracking obligations to ensure patient safety.
- Risk-Based Compliance Adjustments – PMS requirements vary by:
- Device type (general IVDs, high-risk IVDs, etc.)
- Basis of conformity assessment (UKCA, IVDD, or IVDR).
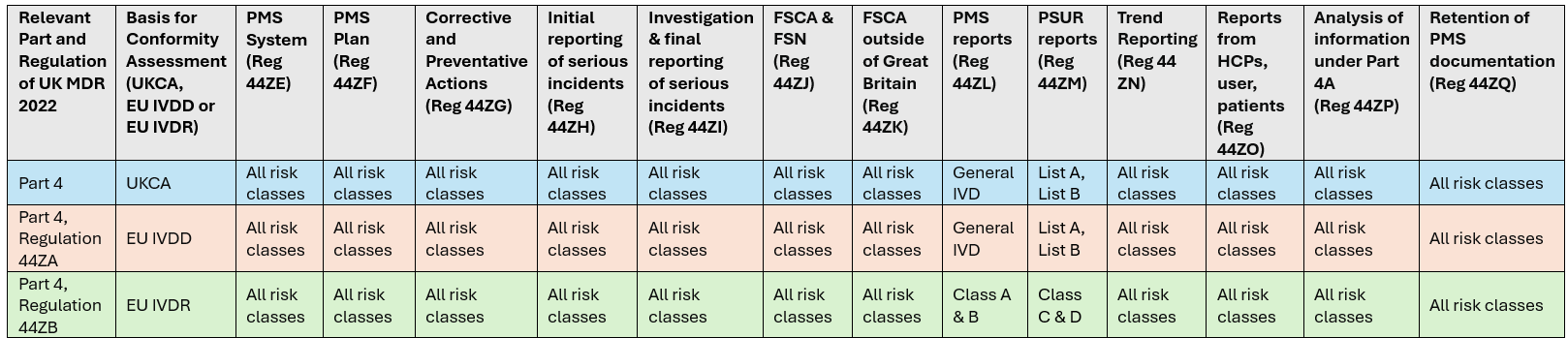
Table 1 reproduces the PMS obligations specific to IVD devices based on conformity assessment[8]. Most requirements are the responsibility of the Manufacturer; however, certain requirements may be delegated to the UK Responsible Person (UKRP)[9].
Exceptions: The new PMS requirements do not apply to:
- Devices subject to clinical investigation, performance evaluation or exceptional use authorisation in GB.
- Devices discontinued before the SI enforcement date (these remain subject to MED DEV 2.12 PMS requirements[10]).
- Devices placed on the market or put into service in Northern Ireland (NI), which must follow the PMS requirements of EU MDR 2017/745 and EU IVDR 2017/746.
The MHRA has published guidance documents clarifying implementation details [11], including stakeholder consultation opportunities through trade associations[12].
Table 1. PMS obligations specific to IVDs* (click to enlarge)
Transitional Arrangements & Industry Impact
Key Transition Deadlines
Regulatory professionals must monitor the following deadlines[13] to ensure continued market access:
- CE-Marked Devices – Transitional allowances until December 31, 2027 for most IVDs (and medical devices), with risk-class specific phase-out dates through 2030.
- UKCA Marking – The UK is shifting towards full UKCA marking adoption, requiring manufacturers to transition accordingly.
- Guidance & Support – MHRA has committed to publishing comprehensive guidance, with further updates expected in 2025.
What Regulatory Professionals Should Do Now
To prepare for the upcoming changes, regulatory professionals should:
- Monitor Implementation Timelines – Stay informed on MHRA consultations, SI publications, and transition deadlines.
- Engage with MHRA Guidance – Regularly review PMS updates, UKCA transition arrangements, and new IVD-specific regulations.
- Conduct regulatory gap analyses – Identify necessary updates to PMS documentation, quality management systems and labeling.
- Collaborate with Industry Peers – Participate in trade associations, regulatory working groups, and discussions with UK Approved Bodies.
- Plan for UKCA Transition – Develop a clear strategy for transitioning from CE to UKCA marking.
Our Expertise in IVD & Medical Device Compliance
At Beaufort, we offer extensive expertise in IVD regulatory affairs, clinical research, quality management, and market access. Our team of regulatory experts can help you:
- Develop & Implement PMS Systems – Ensure compliance with the new UK PMS requirements, including PMS plans, PSURs, and incident reporting.
- Prepare for UKCA Marking – Assist with conformity assessments, documentation updates, and transition planning.
- Regulatory Gap Analysis & Compliance Strategy – Identify compliance gaps and develop gap-closure roadmaps.
- Regulatory Submission Support – Guide you through the MHRA regulatory submission process, including exceptional use authorizations and performance evaluations.
- Performance Evaluation Support – Design clinical and performance evaluation studies to meet new UK and international requirements.
- Training & Consultation – Provide customized regulatory training and consultation.
Final Thoughts
With the UK’s evolving regulatory framework, staying ahead of the MHRA roadmap is critical for IVD manufacturers and regulatory professionals. As a trusted CRO partner, Beaufort is ready to help companies navigate these regulatory changes, maintain compliance, and ensure market access in the UK.
For more information on how Beaufort can support your UK regulatory strategy, contact us directly at [email protected] or provide your contact information here.
[1] Medicines and Healthcare products Regulatory Agency. Guidance Regulating medical devices in the UK. Published 15 January 2025. Accessed February 5, 2025. https://www.gov.uk/guidance/regulating-medical-devices-in-the-uk#NI.
[2] Medicines and Healthcare products Regulatory Agency. Guidance on the regulation of In Vitro Diagnostic medical devices in Great Britain. Published January 2025. Accessed February 5, 2025. https://assets.publishing.service.gov.uk/media/67863a313ef063b15dca0f47/Guidance_on_the_regulation_of_IVD_medical_devices_in_GB.pdf.
[3] Medicines and Healthcare products Regulatory Agency. Guidance for retailers: supplying medical devices to Northern Ireland. Last updated 5 March 2021. Accessed February 5, 2025. https://www.gov.uk/guidance/guidance-for-retailers-supplying-medical-devices-to-northern-ireland#:~:text=A%20key%20part%20of%20the,to%20the%20relevant%20EU%20legislation
[4] Medicines and Healthcare products Regulatory Agency. Government response to consultation on the future regulation of medical devices in the United Kingdom. Published 26 June 2022. Accessed February 5, 2025. https://assets.publishing.service.gov.uk/media/62b577f6d3bf7f0b00165a32/Government_response_to_consultation_on_the_future_regulation_of_medical_devices_in_the_United_Kingdom.pdf. [1] Medicines and
[5] Healthcare products Regulatory Agency. Medical Devices Regulatory Reform Roadmap to implementation. Version 2.0 (December 2024). Accessed February 5, 2025. https://assets.publishing.service.gov.uk/media/6759a8827e419d6e07ce2b21/Med_Tech_Regulatory_Roadmap_V2_December_2024.pdf.
[6] The Medical Devices (Post-market Surveillance Requirements) (Amendment) (Great Britain) Regulations 2024 – UK Statutory Instruments 2024 No. 1368 Assessed February 10, 2025. https://www.legislation.gov.uk/uksi/2024/1368/contents/made
[7] Medicines and Healthcare products Regulatory Agency. Guidance The Medical Devices (Post-market Surveillance Requirements) (Amendment) (Great Britain) Regulations 2024: guidance on implementation. Published 15 January 2025. Accessed February 5, 2025. https://www.gov.uk/government/publications/medical-devices-post-market-surveillance-requirements/the-medical-devices-post-market-surveillance-requirements-amendment-great-britain-regulations-2024-guidance-on-implementation
[8] Medicines and Healthcare products Regulatory Agency. Guidance Post-market surveillance (PMS) obligations by medical device type. Published 15 January 2025. Accessed February 5, 2025.https://www.gov.uk/government/publications/medical-devices-post-market-surveillance-requirements/post-market-surveillance-pms-obligations-by-medical-device-type
[9] Medicines and Healthcare products Regulatory Agency. Guidance Post-market surveillance requirements for medical devices: summary of main changes. Published 15 January 2025. Accessed February 5, 2025. https://www.gov.uk/government/publications/medical-devices-post-market-surveillance-requirements/post-market-surveillance-requirements-for-medical-devices-summary-of-main-changes
[10] Guidance MEDDEVs. Accessed February 5, 2025. https://health.ec.europa.eu/document/download/c1a6aa0b-d8c8-498b-8ed4-9f3c6211896d_en
[11] Medicines and Healthcare products Regulatory Agency. Guidance The Medical Devices (Post-market Surveillance Requirements) (Amendment) (Great Britain) Regulations 2024: guidance on implementation. Published 15 January 2025. Accessed February 5, 2025. https://www.gov.uk/government/publications/medical-devices-post-market-surveillance-requirements/the-medical-devices-post-market-surveillance-requirements-amendment-great-britain-regulations-2024-guidance-on-implementation.
[12] Medicines and Healthcare products Regulatory Agency. Press release. MHRA guidance on new Medical Devices Post-Market Surveillance requirements. Published 15 January 2025. Accessed February 5, 2025. https://www.gov.uk/government/news/mhra-guidance-on-new-medical-devices-post-market-surveillance-requirements
[13] Medicines and Healthcare products Regulatory Agency. Infographic – Timelines for placing CE marked IVDs on the Great Britain market. Accessed February 10, 2025. https://assets.publishing.service.gov.uk/media/6718b88738149ce9d09e3894/Infographic_-_Devices_transition_timeline.pdf
The European Union’s regulatory framework for healthcare innovation is among the most advanced globally, yet its complexity often presents significant challenges. This is especially true for “combined studies” involving medicinal products, medical devices, and in vitro diagnostics (IVDs).
Recognizing these challenges, the EU launched the COMBINE project in June 2023 to streamline the regulatory landscape for such studies to ensure that innovative treatments reach EU patients while maintaining compliance with high standards.
Understanding Combined Studies and Regulatory Complexity
Combined studies, which involve the simultaneous investigation of one or more medicinal products, IVDs, and/or medical devices must comply with multiple regulatory frameworks:
- Regulation (EU) 536/2014 on clinical trials of medicinal products (CTR)
- Regulation (EU) 2017/745 on medical devices (MDR)
- Regulation (EU) 2017/746 on in vitro diagnostic medical devices (IVDR)
Each framework governs distinct yet interrelated aspects of healthcare product development. Divergent requirements, documentation, timelines, and processes across these frameworks pose significant barriers, delaying trial initiation and limiting patient access to innovative therapies.
The Impact of IVDR on Using Companion Diagnostics in Clinical Trials
Companion diagnostics (CDx) are often co-developed together with their medicinal product and are evaluated in combined studies. Previously, under Directive 98/79/EC (IVDD), CDx were not defined and such “biomarker assays” were classified as “IVD other” (not Annex II; low risk). Regulatory oversite for use of biomarker assays in clinical trials occurred at the Member State level and requirements were not harmonized.
With the entry into force of the Regulation (EU) 2917/746, a new risk-based classification system (classes A to D) was introduced and a legal definition for CDx established. Companion diagnostics are Class C devices and when used in clinical trials, are now required to be CE-marked for their intended use in the trial, authorized for in-house use, or developed for clinical performance evaluation.
Clinical performance studies (PS) involving CDx must adhere not only to the general requirements for all PS set out in IVDR Article 57 and Annex XIII, but must also be designed, authorised, conducted, recorded and reported in accordance with Articles 58 to 77 and Annex XIV as for “interventional clinical performance studies” as CDx represent a significant level of inherent risk.
These changes have contributed to significant delays in clinical trial initiation.
A 2023 report by the European Federation of Pharmaceutical Industries and Associations (EFPIA) highlighted that 43% of companies expected delays of 6 to 12 months in starting their trials, with 228 to 410 trials enrolling fewer EU patients. In the context of oncology alone, this could mean delayed access to life-saving treatments for as many as 42,200 patients over the next three years[1].
The COMBINE Project: Finding Solutions to Regulatory Barriers
In response to these growing issues, the COMBINE project established a two-phased approach with the goal of first understanding the encountered challenges and then implementing solutions. This involves cross-functional collaboration at the EU level, including experts from the different EU governance structures. Additionally, an external stakeholder group, representing industry, patients, academic research groups, health care professionals, clinicians and notified bodies contributes to the effort.
Phase 1: Analysis – Mapping the Challenges and Early Insights
The first phase of COMBINE, conducted between September 2023 and May 2024[2], was dedicated to understanding the challenges sponsors face when conducting combined studies. The analysis phase had four key objectives:
- Identifying Issues: Stakeholder workshops revealed 78 unique issues, ranging from misaligned regulatory timelines to unclear definitions and procedural inconsistencies.
- Mapping National Processes: A survey across 24 EU Member States uncovered significant inconsistencies in how national competent authorities (NCAs) and ethics committees (ECs) handle combined studies.
- Reviewing Ongoing Work: Existing EU-level guidance and activities were assessed for areas of alignment and gaps.
- Proposing Solutions: The issues were analyzed, and potential solutions were grouped into thematic areas such as coordinated assessments, alignment, and improved communication.
The analysis phase also identified more than 50 proposed actions to address these issues and concluded with a roadmap that emphasized:
- Establishing coordinated assessments for combined studies to reduce duplication and harmonize processes.
- Aligning Member State interpretations and procedures to minimize variability.
- Enhancing communication between stakeholders through training, dialogue forums, and better IT integration.
Phase 2: COMBINE Programme Vision and Strategic Implementation Plan
In December 2024, the EU Member States endorsed the strategy for the second phase: the “COMBINE programme”.
Building on insights gained from its analysis, the COMBINE Programme implementation phase is designed to deliver solutions for challenges facing combined studies in the EU.
The recently published ‘COMBINE’ programme strategy’ document[3], clarifies the vision, its structure and governance, and provides an overall plan of activities.
The COMBINE programme seeks to make the European Union an attractive region to conduct combined studies, envisioning a clear and smoothly functioning regulatory environment for combined studies through broad involvement of regulators, ethics committees and all impacted stakeholders, with the ultimate goal to support availability of innovative treatments for patients.
Structure and Governance
In the EU, there is no single regulatory authority. Medical devices are governed at a European level by the EU Commission expert groups, organized under the Medical Device Coordination Group (MDCG) while medicines are governed by the Heads of Medicines Agency (HMA), the EU Commission and the European Medicines Agency (EMA).
Further, National Comptent Authority (NCA) clinical trial authorization is governed by the Clinical Trials Coordination Group (CTCG) at the HMA and the Clinical Trials Coordination and Advisory Group (CTAG) at the EU Commission. And the Ethics Committees system is made up of national Research Ethics Committees (RECs) for both medicines and devices, sometimes the same Committees cover both aspects and sometimes the Committees are separate.
This affords unique challenges in solving issues with cross-sectorial relevance. As such, the COMBINE programme aims to provide a framework for cross-sectorial work in relation to combined studies that cannot be solved by a single sector and is not intended to replace or significantly change already existing structures.
Programme Action Plan Highlights
The programme’s activities are organized into three thematic areas:
1. Coordinated Assessments
This pillar focuses on harmonizing the evaluation of combined study applications by competent authorities, as well as by ethics committees by:
- Exploring coordinating assessments of applications for combined studies among Member States (including competent authorities and ethics committees) and across the CTR and IVDR or MDR.
- Piloting a coordinated assessment procedure for clinical trials involving IVDs as companion diagnostics.
- Integrating learnings into ongoing work on integration between the CTR Clinical Trials Information System (CTIS) and the MDR/IVDR European database on medical devices (Eudamed).
2. Alignment
Efforts under this area aim to align interpretations and procedures across the EU Member States by:
- Providing an aligned approach on the processes for combined studies (e.g., same IVD used in multiple IMPs, procedures for substantial modifications).
- Optimizing serious adverse event reporting in combined studies.
3. Communication and Dialogue
This thematic area emphasizes improving stakeholder exchange of information, advice and training, including:
- Enhancing training for sponsors and assessors.
- Fostering member state collaboration through forums and best practice exchanges.
A Phased Implementation Approach (2024-2027)
To ensure feasibility and effective resource allocation, the action plan is divided into three stages:
- Stage 1 (2024 – Q1 2025): Focus on coordinated assessments and initial alignment activities.
- Stage 2 (Q2 2025 – Q1 2026): Expand alignment efforts and initiate communication and dialogue-focused projects.
- Stage 3 (Q2 2026 – Q1 2027): Sustain communication and dialogue initiatives.
The Road Ahead
The COMBINE project provides a roadmap to address regulatory inefficiencies, ensuring combined studies can progress without unnecessary delays. Its success depends on sustained collaboration and commitment across the EU regulatory ecosystem. As the EU continues to refine its approach to combined studies, the lessons learned, and solutions implemented through COMBINE will serve as a blueprint for future regulatory innovations.
These solutions are essential to reducing the regulatory burden on sponsors, speeding up trial initiation, and ensuring that European patients have access to cutting-edge therapies.

Beaufort’s Commitment to Innovation and Patient Access
As regulatory frameworks evolve, Beaufort’s expertise positions us as a trusted partner for sponsors navigating the complexities of combined studies. By staying abreast of regulatory changes and advocating for streamlined processes, we help our clients succeed and ensure that patients benefit from timely access to innovative treatments. Beaufort remains committed to driving excellence in regulatory strategy, fostering a future where innovation and compliance go hand in hand.
Contact us to learn more about how Beaufort can successfully support your current or future clinical trial.
[1] https://health.ec.europa.eu/document/download/77e1409a-f4c0-45db-bff1-4873c7a0e7ae_en?filename=md_combined-analysis-phase-report_0.pdf
[2] European Federation of Pharmaceutical Industries and Associations. (2023). Critical Impacts of IVDR Implementation on Patient Access to Clinical Trials. https://efpia.eu/media/677143/efpia_ivdr-survey-slides.pdf
[3] https://health.ec.europa.eu/document/download/c10c325f-ae88-4956-a7eb-f45acc0a9811_en?filename=md_combine_strategy_en.pdf
On January 23, 2024, the European Commission posted a Proposal for a further extension to companies to apply the In Vitro Diagnostic Medical Devices Regulation (IVDR), under certain conditions. Specifically, this Proposal suggests granting a new extended timeline for companies with IVD products already on market in the EU depending on the risk classification of the device. There will be a shorter transition period for high risk IVDs and longer periods for medium and lower risk IVDs, as follows:
- high individual and public health risk devices such as HIV or hepatitis tests (class D) would have a transition period until December, 2027;
- high individual and/or moderate public health risk devices such as cancer tests (class C), would have a transition period until December, 2028;
- lower risk devices (class B such as pregnancy tests and class A sterile devices such as blood collection tubes), have a transition period until December, 2029.
The Proposal is not final and will now be put forward to the European Parliament and Council.
If adopted, only ‘legacy devices’, meaning devices covered by a certificate or declaration of conformity issued under the previous legal framework (notably Directive 98/79/EC), may benefit from the extended transition periods. And, as with the currently in effect transitional provisions, only if they continue to comply with the rules in force when they were placed on the market for the first time and if there are no significant changes in the design or intended purpose of the devices;
However, this Proposal introduces the following additional conditions in order for legacy devices to benefit from the extended timelines:
- the devices do not present an unacceptable risk to the health or safety of patients, users or other persons, or to other aspects of the protection of public health;
- no later than 26 May 2025, the manufacturer puts in place a quality management system compliant with the IVD Regulation;
- for devices requiring an assessment by a notified body, the manufacturer submits an application to the notified body to transfer the device to the IVD Regulation by 26 May 2025 (class D), 2026 (class C) or 2027 (class B and A sterile IVDs). The manufacturer and the notified body sign a written agreement to proceed with conformity assessment shortly after those dates.
The above new proposed requirements may mean that those with class C or class B and class A (sterile) IVDs have less time in which to put in place an IVDR-compliant quality management system
Thus, while the Proposal buys those with CE-marked IVDs already on market more time, the above new proposed requirements may mean that those with class C or class B and class A (sterile) IVDs have less time in which to put in place an IVDR-compliant quality management system and less time to submit an application to their Notified Body to transfer legacy devices to the IVD Regulation by 26 May 2025 (class D), 2026 (class C) or 2027 (class B and A sterile IVDs). This last condition aims to ensure that only devices that the manufacturer intends to transition to the IVDR will benefit from the extended transition period.
For companion diagnostic (CDx) companies, non-CE marked CDx will need to move forward as planned. However, the extended timelines could alleviate issues with Notified Body capacity if they strive to get their IVDs through the conformity assessment process ahead of the revised transitional provisions, keeping in mind the shorter timeline for Class D devices.
Additionally, not providing immediate relief, the Proposal also calls for the timelines for the application of the coordinated assessment of performance studies (IVDR Article 74) to be adapted. Keeping the approach provided for in the IVDR, the Proposal indicates the coordinated assessment procedure for performance studies should first be applied by Member States on an ‘opt-in’ basis but five years after its voluntary application, the coordinated assessment should become mandatory for all Member States. If adopted, these changes should ultimately aid in offsetting the delay in the development of the Eudamed module for performance studies. Those companies involved in combined studies (i.e., those a clinical trial of a medical product in parallel with a performance study of an in vitro diagnostic) may reap the greatest benefit.
We are also keeping abreast of the developments of the COMBINE project, launched in June 2023. The Member State’s competent authorities for clinical trials and medical devices and the European Commission initiated the COMBINE project to analyse the root causes of the challenges encountered by sponsors in conducting combined studies and to identify possible solutions to these challenges.
The Proposal also calls for mandatory use of some European database on medical devices (Eudamed) modules that are already finalized, and new obligation for manufacturers to give prior notice in case of disruption of supply of certain IVDs.
The EU Commission’s press release and links for associated documents can be reviewed here: https://lnkd.in/g_Es65ku
The full Proposal can be viewed here: https://lnkd.in/gA7wA4JS
The EU Commission’s overview of the COMBINE project can be viewed here: https://health.ec.europa.eu/medical-devices-topics-interest/combined-studies_en
How We Can Help
Beaufort’s team of experts are currently helping companies navigate the continually evolving IVDR landscape. We have on-going projects focusing on QMS implementation, Technical Documentation preparation and review and navigation of the current processes for preparation and review of National requirements for companion diagnostic clinical performance study documentation and Competent Authority/EC review.
Contact us today for a consultation.
Key Takeaways for IVD manufacturers
Authored By: Karin Hughes, SVP Global Regulatory and Quality
While the European Union (EU), United Kingdom (UK) and Switzerland once presented a largely harmonized and favorable first market for launching diagnostic products, the current era, post-Brexit, post-Swixit and a year post the implementation date of the EU In Vitro Diagnostics Regulation (IVDR) presents in vitro diagnostic (IVD) manufacturers with enormous challenges. Extensive and more stringent conformity assessment requirements have emerged, most notably through the application of the IVDR (Regulation (EU) 2017/746) and, in some regimes (i.e., the UK), new Medical Device Regulations are not yet published.
While the primary aim of the IVDR – which became effective on May 26, 2022– and the future UK Medical Device Regulations is to ensure better protection of public health and patient safety, the resulting costs, and complexities of introducing new products to these markets, as well as maintaining IVD legacy products on market, have increased dramatically.
The June 2023 MedTech Summit in Brussels, Belgium, offered a number of insightful updates on the evolving European and UK regulatory landscapes. Participants included several Notified Bodies (TÜV SÜD, TÜV Rheinland, BSI and DEKRA), and UK representatives from the British In Vitro Diagnostic Association (BVDA) and the Association of British HealthTech Industries (ABHI). Participants also included national competent authorities (NCAs) from Germany (Paul-Ehrlich-Institut), Finland (Finnish Medicines Agency – Fimea), and Ireland (National Standards Authority of Ireland), as well as those of us responsible for the implementation of these requirements.
Here, we report some of the most salient insights for IVD manufacturers, including:
● The Ebb and Flow of Notified Body Capacity
● Updates on Companion Diagnostics
● European Database on Medical Devices (EUDAMED) Update
● The Changing Landscape for Distance Sales
● COVID-19 (SARS CoV and SARS-CoV-2) Classification
● Clinical Performance Data Requirements
The Ebb and Flow of Notified Body Capacity
More than one year after the implementation of the IVDR — replacing the In Vitro Diagnostic Medical Devices Directive 98/79/EC (IVDD) and related legislation of individual EU nations — several issues and challenges have emerged. Yet, despite the shortcomings, positive news reported at the MedTech Summit is that more IVD manufacturers are successfully navigating the process.
One of the larger challenges facing the system has been the lack of sufficient Notified Body (NB) capacity. Under the old directive, the number of IVD devices that required an NB conformity assessment was approximately 20 percent. Now, under the IVDR, that number has grown to roughly 80 to 90 percent.
Notified Bodies have been navigating their own processes under the IVDR to (1) become designated to perform conformity assessments, and (2) to expand the scope of their Product Families, Procedures and Articles/Annexes for which they have demonstrated competence. Additionally, a large number of IVD manufacturers have required extensions of IVDD certificates prior to the 2022 date of application. The combination of these three factors has consumed NB capacity over the past year, and, as recently as six months ago, several NBs were unable to accept new applications.
On the upside, those NBs represented at the MedTech Summit reported that their capacity has greatly improved, and the number of certificates that were issued under the IVDR tripled in the second half of 2022. All summit-participating NBs are currently accepting new applications. As well, two companion diagnostics (CDx) have been issued certificates — one by TÜV SÜD and another by BSI. What’s more, at least six European Medicines Agency (EMA) consultations have been completed, with more in the pipeline.
Notified Bodies are urging manufacturers to continue moving forward with their applications and assessments. With the implementation of the IVDR transitional provisions for IVDs (those previously certified for the European market in compliance with the IVDD), representatives from various NBs noted that they have begun to see application withdrawals and a decline in industry submissions for conformity assessments. As a result, NBs are concerned that as industry delays conformity assessment submissions, a new capacity challenge may result. As transitional provisions bring Class D submissions online (due by May 26, 2025), there is a risk the timing may coincide with a new push by IVD manufacturers to attempt to get their IVDs reviewed under the IVDR.
The timetable for successfully receiving certification does not favor delaying submission for conformity assessments. Prior to review, it may take several months to complete the request-for-quote process, and have an NB subsequently assign appropriate subject matter resources to begin the technical documentation review. Participating NBs noted that, on average, conformity assessments are still taking one year to complete, with an observed range of nine to 24 months. As well, on the back end, there can be as much as another three to six months after a device has been recommended for certification to actually receive the certificate. For those who may have had experience with CE-marked List II A or self-test IVDs under the IVDD, these IVDR review timelines still exceed those six-month reviews common under the IVDD. Dedicated review – NB service wherein IVD manufacturers pay an extra fee (per conformity assessment review) for a focused, dedicated review – may provide shorter timelines, but can be costly. Proper device classification and NBOG code assignment and high-quality submissions, which meet all applicable Technical Documentation requirements and include justifications for those not applicable, remain the best timeline risk mitigations..
Updates on Companion Diagnostics
For companion diagnostics (CDx), two factors — (1) the delay in the availability of the coordinated assessment procedures for the review and approval of CDx performance study applications, and (2) the current lack of harmonization in national legislation requirements for NCA and ethic committee application documentation and reviews — are continuing to negatively impact the ability to initiate clinical trials of investigational medicinal products (CTIMPs) that rely on a CDx for medical management decisions. While the European Medicines Agency (EMA) offers scientific and protocol assistance to developers of medicinal products through multiple pathways — including the Simultaneous National Scientific Advice (SNSA) pilot, which has been successfully used by CDx developers in conjunction with their pharma partners — the MedTech Summit CDx panel noted there is no process for structured dialog among all stakeholders (e.g., medicinal product developer, diagnostics manufacturer, NB and EMA) prior to, or during, the conformity assessment of the CDx and the medicinal product authorization. This lack of guidance puts at risk the simultaneous approval of the medicinal product, alongside certification of the CDx, potentially delaying timely access for patients to both
There was welcome news that the EMA has established a “focus group on provision of scientific advice for medicinal product developments comprising drug-device combinations and drug-CDx combinations.” This is meant to be a broader exchange platform for EMA, Scientific Advice Working Party (SAWP) members, NCA experts, NBs, and industry. The initial goal is to understand the perspectives of the stakeholders through case studies to determine when, where and from whom advice is needed. This group is also exploring the possibility of structured exchanges between stakeholders.
The kickoff meeting occurred two months before the MedTech Summit, and there have been four meetings to date, with the results of these meetings expected to be published by the EMA. It was also noted that there is on-going activity by the EMA’s Methodology Working Party aimed at providing guidance for simultaneous medicines authorization and CDx certification based on the EMA/Committee for Medicinal Products for Human Use 2016 Concept paper on predictive biomarker-based assay development in the context of drug development and lifecycle. The working party is moving to finalize the guideline as soon as possible.
European Database on Medical Devices Update
One year after IVDR implementation, the regulatory infrastructure remains incomplete and continues to pose a challenge. The European Database on Medical Devices (EUDAMED) had been scheduled to go live in May 2020, but, as yet, has achieved only half of its implementation goals. Composed of six modules — actor registration; unique device identification (UDI) and device registration; NBs and certificates; clinical investigations and performance studies; vigilance; and market surveillance — EUDAMED is intended to provide a “living picture” of the lifecycle of medical devices that are made available in the EU.
The EUDAMED production environment currently contains three modules — actor registration, UDI and device registration, and NBs and certificates — containing valid data for devices placed on the EU market. Significant progress continues to be made towards full implementation in Q4 2024, and the European Commission (EC) continues to target Q2 2024 for the remaining modules to be available.
Once EUDAMED has achieved full functionality, and there is publication of an EC notice in the Official Journal of the European Union (OJEU), industry will have six months before use of EUDAMED becomes mandatory with respect to the obligations related to actors, vigilance, clinical investigations and performance studies, and market surveillance modules. Twenty-four months after publication of the notice in OJEU (Q2 2026), the use of EUDAMED becomes mandatory with respect to the obligations related to UDI and device registration, NBs, and certificates.
The Changing Landscape for Distance Sales
Distance sales — non-EU-based clinical laboratories providing services to EU patients — lack guidance under the new IVDR, and many U.S. laboratory-developed test (LDT) providers are unaware of the new requirements. Or, if aware, they may incorrectly believe their testing is exempt under the “in-house” exemption provisions described in Article 5 of the IVDR, which allow healthcare institutions to manufacture, modify, and use in-house tests on a non-industrial scale to meet the specific needs of target patient groups, if an equivalent device available on the market cannot already meet these needs at the appropriate level of performance.
However, this exemption applies only to labs that are part of healthcare institutions established in the EU. All non-EU and most EU-based commercial laboratories will, therefore, be required to CE mark their LDTs. For the subset of labs that meet the in-house exemption, the IVDR will still require them to meet several new standards, including compliance with the IVDR’s Annex I – General Safety and Performance Requirements and quality management system framework.
While the requirements for distance sales are found in Article 6 of the IVDR, they are brief and not straightforward. NBs at the MedTech Summit noted that it has been a challenge for them to interpret those requirements. One NB noted that manufacturers often do not realize they are performing distance sales, and the NB only uncovers this during technical documentation review. Also, from an NCA point of view, Article 6 presents uncertainty. For example, if a U.S. company offers laboratory services via the internet, accessible by anyone in the EU, which country/NCA has responsibility for review?
On a positive note, a distance sales task force has been initiated. However, the Medical Device Coordination Group (MDCG) guidance and/or a Q&A paper is not imminent; any results from the task force will be eagerly awaited by manufacturers, with hope for a clear implementation pathway for distance sales requirements.
COVID-19 (SARS CoV and SARS-CoV-2) Classification
With the World Health Organization downgrading the COVID-19 pandemic, and stating it is no longer qualified as a global emergency in May of this year, IVD manufacturers of SARS-CoV-2 diagnostics are looking for a decision as to whether these products will be down-classified under the IVDR.
These discussions have reached the EC, who is consulting NCAs. Currently, in MDCG 2020-16 rev.2, SARS-CoV and SARS-CoV-2 are listed as examples of products classified as Class D under Rule 1 as a transmissible agent. The MDCG 2020-16 has already undergone revision, and down-classification would minimally require additional changes to guidance. However, some manufacturers are choosing not to wait, and are undertaking a classification dispute with their NB. In accordance with IVDR Article 47, such classification disputes are referred for a decision to the particular NCA. The NCA then notifies the MDCG and the EC of its decision. SARS-CoV-2 tests are currently trending at 42 percent of Class D by application.
Clinical Performance Requirements
Clinical performance requirements for IVD certification remain an issue, particularly for legacy devices. Demonstration of clinical performance for legacy devices is often based on studies conducted under the IVDD, and IVD manufacturers may have difficulty justifying suitability of their products’ performance with that of the generally acknowledged state of the art in medicine. IVD manufacturers may not have the institutional or financial support to conduct new performance studies to fulfill gaps to meet the more burdensome clinical performance requirements.
While the NBs are aware of the challenges of legacy devices, manufacturers must justify whether using previous data, without having to produce any new data, is sufficient to support their intended purpose. It may be possible to augment previously conducted performance evaluation data with literature and/or data from published experience gained by routine diagnostic testing. Otherwise, a change to the intended purpose, resulting in limited and less desirable claims, or market removal may be necessary. Deficiencies in performance evaluation continue to be the number one gap in Technical Documentation cited by the NBs for legacy devices.
Navigating the New IVDR With Beaufort
The transition from the IVDD to the IVDR continues to be challenging. Lack of the expected infrastructure (e.g., EUDAMED, EU Reference Labs, etc.) coupled with the slow pace of available guidance still requires evolutionary steps before solidifying into a cohesive and comprehensive regulatory regime. While not unexpected, the impact to stakeholders remains high, and continues to add risk to commercialization efforts.
Beaufort’s expertise can help you effectively address your IVDR challenges from Covid to Companion Diagnostics. Our team is assisting clients from implementation of quality system requirements and establishment of clinical evidence to technical writing of reports and final Technical Documentation. We are also constantly monitoring the continuously changing regulatory landscape so we can effectively consider near-term and evolving cross-regulatory-regime requirements when developing regulatory strategies to reduce the overall pre-market burden across the US, EU, UK, and ROW markets.
We bring all the pieces together to create a clear and actionable roadmap for legacy and new products to meet the IVDR requirements and adjust that roadmap to meet emerging changes in the IVDR infrastructure and newly published Guidance.
Contact us today to see how we can help you bring your IVD product successfully through the changing regulatory landscape.
As we move closer to the May 2022 deadline where all IVD products marketed in the EU must meet the new IVDR regulations, it is imperative for manufacturers to begin the process of implementing the mandated requirements.
To help manufacturers navigate this complex landscape, Karin Hughes, PhD, Senior Vice President, Regulatory & Quality, delivered a presentation during the 14th Annual IVD Clinical & Regulatory Affairs Conference in October, 2020. This webinar, Ensuring Compliance with IVDR Clinical Requirements, covers considerations and best practices across the following topics:
- Developing clinical evidence reflective of a European population
- Establishment of scientific, clinical, & analytical validity
- Implementation of a risk based post market surveillance system
- Confirming the IVD test adheres to “state of the art” definition
WATCH WEBINAR
Beaufort can support all facets of your IVDR implementation. Our team of IVDR subject matter experts have an in-depth understanding of the complex details, regulatory expectations and new mandatory requirements. We provide the strategic and technical guidance to develop an appropriate project plan, as well as assist with the implementation of any stage of the IVDR certification process.
Read more about Beaufort’s comprehensive IVDR compliance services. Interested in having a conversation? Email us at [email protected] or submit your request today.
Beaufort CRO was proud to sponsor the 14th Annual IVD Clinical & Regulatory Affairs Conference in October, 2020, and partner with Q1 Productions on this important industry event.
The long-awaited Medical Device Coordination Group (MDCG) Guidance on Clinical Evidence is now available.
The 31-page document, MDC 2022-2, outlines how clinical evidence is to be demonstrated pursuant to the requirements of the IVDR.
What does this mean for study sponsors and manufacturers? In short, the need for continuous oversight of clinical data and performance evaluation throughout the IVD’s entire lifecycle from pre-market release to post-market surveillance and follow-up.
The guidance, which also includes more than 5 pages of definitions, details the specific principles and procedures to follow over the entire lifetime of a product, with dedicated sections across the following areas:
- General principles of Clinical Evidence
- Performance evaluation process
- The role of risk management in performance evaluation
- Performance Evaluation Plan (PEP)
- Scientific Validity, Analytical Performance and Clinical Performance
- Performance Evaluation Report (PER)
- Continuous update of the performance evaluation
The last point on continuous performance evaluation reflects the overarching approach manufacturers must adopt, as stated in the excerpt below:
“Scientific developments and improvements in state-of-the-art should be reviewed and assessed by the manufacturer on a regular basis as part of their continuous and pro-active postmarket surveillance activities. Therefore, manufacturers must instate a procedure for planned monitoring of scientific developments and changes in medical practice relevant to the IVD(s). Any relevant new information, developments and progress in the scientific field should trigger reassessments of the existing clinical evidence thus ensuring safety and performance through a continuous performance evaluation process.”
This guidance document also includes an appendix on the methodological principle for generating clinical evidence as well as the required frequency for updating reports. All of which reinforces the critical need for a systematic approach to short and long-term study planning – and the benefit of having Beaufort’s highly-skilled IVDR team by your side.