

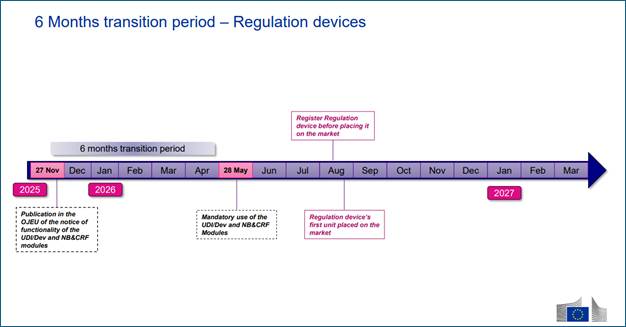
On 27 November 2025, the European Commission published Commission Decision (EU) 2025/2371, formally confirming that the first four modules of EUDAMED…
Authored By: Karin Hughes, Ph.D., SVP Global Regulatory & Quality
On 27 November 2025, the European Commission published Commission Decision (EU) 2025/2371, formally confirming that the first four modules of EUDAMED—Actor Registration, UDI/Devices, Notified Bodies & Certificates, and Market Surveillance—have achieved full functional compliance under the Medical Devices Regulation (EU) 2017/745 (MDR) and the In Vitro Diagnostic Medical Devices Regulation (EU) 2017/746 (IVDR).
Publication of the notice in the Official Journal of the European Union (OJEU) triggered the 6-month transition period under Regulation (EU) 2024/1860, making 28 May 2026 as the mandatory-use date for these modules.
What is EUDAMED?
EUDAMED — the European Database on Medical Devices — is the European Commission’s centralized, interoperable IT system that supports the operational implementation of the MDR and IVDR.
EUDAMED is designed to:
- Improve transparency of medical devices and IVDs on the EU market
- Enhance traceability across the device lifecycle
- Strengthen oversight by competent authorities and notified bodies
- Support comprehensive post-market surveillance
- Serve as the EU’s unified regulatory information source for stakeholders, including the public
EUDAMED consolidates the regulatory lifecycle—from actor registration, device identification, certification status, and (in future) clinical study and post-market surveillance and vigilance reporting—into a single environment.
Why EUDAMED Matters for IVD Manufacturers
For IVD manufacturers operating under the IVDR, EUDAMED is the primary system through which compliance will be demonstrated and monitored. Its function extends beyond device registration; EUDAMED becomes the central reference point linking pre-market information to post-market obligations.
Key implications include:
- New IVDs placed on the market after 28 May 2026 must be registered in the UDI/Device module, before first placement on the market, with complete and accurate Basic UDI-DI, UDI-DI, intended purpose, classification, device description and manufacturer details.
- Notified-body certificates must be uploaded and continuously maintained within EUDAMED, reflecting real-time certificate status (e.g., amendments, suspensions, withdrawals.
- Legacy IVDs, still legally available under the IVDD/IVDR transitional provisions, must be registered within the transitional deadlines to remain on the market.
- Once the CI/PS and Vigilance/PMS modules are declared functional, key regulatory information including performance study registrations, substantial modifications, summary information, serious incident report and related PMS submissions – will be recorded in EUDAMED.
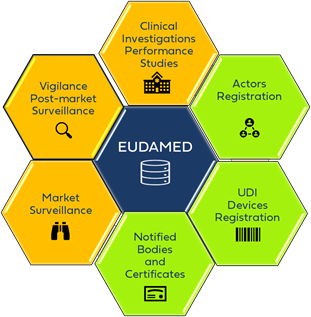
EUDAMED Architecture
EUDAMED consists of a public website and six interconnected modules.
Public Website (EUDAMED Public Interface)
The public interface provides selected transparency into:
- Registered economic operators
- Device and UDI information
- Notified-body certificates
- Required safety or performance summaries.
Not all module information will be public, but transparency is a core design objective of MDR/IVDR.
The Six EUDAMED Modules
1. Actor Registration Module
Captures and maintains information on all economic operators (EOs), i.e., manufacturers, authorized representatives (ARs), importers, and system/procedure-pack producers. Each EO receives an Actor ID, which also functions as a Single Registration Number (SRN) for manufacturers, ARs and importers. The SRN is required before a device or certificate can be registered and is used consistently across EUDAMED.
2. UDI / Device Registration Module
The cornerstone of device traceability, this module records:
- Basic UDI-DI
- UDI-DI
- Device master data
- Classification
- Intended purpose
- Trade names
- Device-family structures
The module supports both initial market access and post-market lifecycle management, ensuring alignment with technical documentation, labeling, and notified-body certification.
3. Notified Bodies & Certificates Module
A restricted-access module used by NBs, their designating authority, competent authorities and the Commission. This module houses medical device/IVD certificates and lifecycle changes (amendments, limitations, suspensions, withdrawals). Certificate records link directly to device registrations.
4. Clinical Investigations & Performance Studies (CI/PS) Module
The CI/PS module remains under development. Once functional, this module will support:
- Registration of clinical investigations and performance studies
- Submission of substantial modifications
- Study-related reporting during study conduct
- Summary results for applicable studies
This module will eventually centralize the regulatory lifecycle for MDR clinical investigations and IVDR performance studies.
5. Vigilance & Post-Market Surveillance (PMS) Module
Also under development. When fully functional, the module will serve as the EU’s centralized environment for post-market regulatory obligations including:
- Serious incident reporting, including initial and follow-up reports submitted by manufacturers
- Trend reporting, where manufacturers identify statistically significant increases in non-serious incidents
- Field Safety Corrective Actions (FSCAs) and corresponding Field Safety Notices
- Periodic Safety Update Reports (PSURs)
- Safety and Clinical Performance Summaries (SSCPs) for certain IVD classes
The PMS module will enable competent authorities to coordinate evaluations, request additional information, and track corrective or preventive actions, making it a crucial component of EU-wide post-market oversight once mandatory use takes effect.
6. Market Surveillance Module
Used primarily by competent authorities and the European Commission, the module supports:
- Inspection planning and reporting
- Surveillance findings
- Enforcement actions
- Inter-authority coordination
This module facilitates harmonized market oversight across the EU.
Modules: What Is Live, What Is Coming (Status December 2025)
| Module | Status | Comments |
| Actor Registration | ✅ Live | Mandatory use beginning 28 May 2026 for all relevant economic operators. |
| UDI / Device Registration | ✅ Live | Mandatory for new devices placed on the market after 28 May 2026. Legacy devices must be registered within the transitional timelines that follow the functionality notice |
| Notified Bodies (NBs) & Certificates | ✅ Live | NBs must upload newly issued MDR/IVDR certificates and all updates as of 28 May 2026. Existing legacy certificates supporting devices that continue to be placed on the market under the MDR/IVDR transitional provisions must be uploaded by the NB within its transitional upload period (up to 18 months following the functionality notice). |
| Market Surveillance | ✅ Live | Mandatory use by competent authorities beginning 28 May 2026. |
| Clinical Investigations & Performance Studies | ⚠️ Pending | Mandatory use only after the Commission publishes a functionality notice in the OJEU. |
| Vigilance & Post-Market Surveillance (PMS) | ⚠️ Pending | Mandatory use only after the Commission publishes a functionality notice in the OJEU. |
Legacy Devices
Regulation (EU) 2024/1860 introduces a phased approach to EUDAMED implementation. For legacy devices—CE-marked under the IVDD, MDD, or AIMDD and legally on the market under transitional provisions—manufacturers must apply the following principles:
- Basic UDI-DI or legacy Basic UDI-DI equivalents apply where UDI-DI obligations do not apply under the Directives (per MDCG 2022-8).
- Economic operators must hold a valid Actor ID/SRN.
- Notified-body certificates supporting legacy IVDs must be uploaded within the NB’s transitional upload period.
- Devices no longer placed on the market after the mandatory-use date do not require registration unless a vigilance or PMS event (e.g., FSCA or serious incident) triggers the need for a minimal device record.
This structured, staged approach allows manufacturers to prioritize registration activities across their portfolios based on commercial relevance and lifecycle status.
Implications for IVD Developers
Pre-Market Readiness
New IVDs placed on the market on or after 28 May 2026 must have:
- A registered economic operator (Actor ID/SRN)
- Completed UDI/Device registration
- Uploaded and current NB certificate information (as applicable)
Legacy Device Management
Legacy IVDs—meaning devices originally CE-marked under the IVDD and those placed on the market under the IVDR transitional provisions—must be registered in EUDAMED using the appropriate Basic UDI-DI or UDI-DI, following the principles in MDCG 2022-8 and within the applicable transitional timelines.
Devices no longer placed on the market may be exempt from EUDAMED registration unless a PMS or vigilance report is required.
Data and Documentation Integrity
Because EUDAMED links:
- Actors
- Device identifiers
- Device master data
- Labeling
- Certification
- PMS information
inconsistencies will be immediately visible. Alignment across QMS processes, technical documentation, UDI structures, and labeling is essential.
Compliance Strategy
With the CI/PS and Vigilance/PMS modules still pending functionality notices, manufacturers should prepare internal processes and procedures now for:
- Performance-study (PS) and clinical-investigation (CI) submissions, along with CI/PS lifecycle modifications
- PSURs, SSCPs, FSCAs and trend reporting
- Integration of digital PMS pathways
Early operational alignment will reduce disruption when these modules become mandatory.
What Sponsors Can Do Next
EUDAMED’s transition to mandatory use reflects a shift toward structured, lifecycle-based regulatory management. As use of the first four modules becomes mandatory and the remaining modules are activated, sponsors will need to ensure that the information supporting their IVDs is accurate, aligned, and updated whenever products, certificates, or economic operators change.
This means viewing EUDAMED as an ongoing part of product lifecycle activities—not a single registration event. Device identifiers, master data, NB certificates, and (in future) CI/PS and PMS information must be maintained in a way that reflects current product status. Organizations that integrate EUDAMED requirements into their routine regulatory and quality processes will be better positioned to manage changes smoothly and avoid downstream issues.
How Beaufort can Assist
Beaufort supports sponsors in building the systems and practices necessary for sustained EUDAMED compliance. Support may include operationalizing EUDAMED requirements by establishing compliant procedures, clarifying roles and responsibilities, and integrating lifecycle needs into existing regulatory and quality systems. Our work extends to supporting documentation for legacy-device registration, preparing internal processes for future CI/PS and Vigilance/PMS modules, and ensuring that regulatory, clinical, and quality activities remain aligned as products evolve.
Whether sponsors are introducing new IVDs, managing transitional portfolios, or preparing for upcoming EUDAMED modules, Beaufort provides the expertise and hands-on support needed to reduce risk, avoid rework, and maintain continuity as EUDAMED becomes central to MDR/IVDR lifecycle management.